Stillbirth continues to cause untold grief throughout the world with the CDC estimating 24,000 stillbirths a year in the United States. The NIH has funded major initiatives over the last couple decades with new information particularly about risk factors1-5. Despite this effort, reducing the number of stillbirths has proven difficult. The persistent high incidence of stillbirth is evidence that there is still a place for new approaches to investigating the causes of stillbirth and finding new opportunities for intervention.
The definition of stillbirth today has to be expanded to that of an infant born without signs of life and who is not successfully resuscitated. Even this definition creates a gray area of some response during resuscitation. The definition of a stillbirth usually has a limit of gestational age. Noting that all fetal loss is important, this invitation to a study will follow the convention that a stillbirth must be at least 20 weeks of gestation.
This invitation for the study of stillbirth is the result of my accumulated autopsy experience. Unlike an epidemiologist who demonstrates an association of a disease, such as maternal diabetes or obesity, with an increased risk of stillbirth, the pathologist attempts to explain why a particular infant of a mother with diabetes or obesity died. The pathologist uses an inductive approach starting with the anatomic evidence and clinical history to create a logical chain of hypotheses about the mechanisms that led to the death. There are no a priori methods to direct the creation of those hypotheses, but they must be consistent with current medical knowledge. In aggregate, reasoning from autopsy data has historically contributed to the understanding and treatment of disease.
This study invitation will concentrate on a subset of stillborn infants in which the pathologist cannot construct a complete chain of causation for the fetal death. This invitation will argue that the anatomic evidence from such autopsies does suggest hypotheses about the cause of death in such stillbirths, and that these hypotheses can be tested. The argument for this study starts with two anatomic finding clusters (Fig 1). The first is a dilated heart and moderate pleural and pericardial effusions. The second is that of petechial hemorrhages on the surface of the intrathoracic organs.

The effusions and dilated heart are classical findings of heart failure generated by incomplete cardiac contraction and increased filling pressures. To some extent, venous congestion in visceral organs is another reflection of these findings. An interpretation of the findings is that they are the result of relatively brief cardiac failure, but longer than expected in sudden cessation of cardiac contractions. The findings do not reflect prolonged heart failure in the fetus which results in anasarca (fetal hydrops) and large pleural effusions often with pulmonary hypoplasia.
The intrathoracic petechiae are focal, not part of generalized thrombocytopenia or consumption coagulopathy as in septic shock. Focal petechiae usually occur from an increase in distending venous blood pressure as with the diagnostic blood pressure cuff test for scurvy that produces petechiae in the lower arm. For intrathoracic petechiae, there is a direct model in Sudden Infant Death Syndrome (SIDS) which often shows thymic petechiae. The current understanding of SIDS is that the infant exhales and then on trying to inhale, the airway is blocked by a foreign object like a blanket. The infant responds by gasping that generates a large negative pressure in the intrathoracic space. This both stops venous outflow from the thorax increasing venous pressure, and likely causes bulging of capillary surfaces into the intrathoracic space. The end result is ruptured capillaries, i.e. petechiae, on the thoracic organ surfaces.
Then by analogy, the intrathoracic petechiae found in stillborn infants are due to the fetus gasping in utero. To complete the analogy, there needs to be an equivalent to the obstructing blanket. In the newborn, in order to overcome the initial atelectasis of the collapsed lung, there is a reflex glottic obstruction, that forces the fetus to develop a strong negative intrathoracic pressure to aid the first breath. It is this glottic stop that provides the analogy to the obstructing blanket. There still needs to be an explanation of why the fetus, whose respirations are normally shallow, suddenly gasps as if trying to take a first breath. The answer can be seen in the results of early experiments of birth asphyxia in newborn monkeys. In the experiments, a plastic bag was placed over the head of a newborn monkey blocking the air way. As can be seen on the graph (Fig 2) from Dr. Dawes monograph, at first the monkey does not take a breath (primary apnea), but as blood oxygen falls, the monkey starts to gasp and continues to do so as the pulse and blood pressure fall over the first 10 minutes of life6. At that point, the infant enters a second stage of apnea and would have died if not resuscitated. This is the model of complete asphyxia. A fetus with complete occlusion of the umbilical cord, or complete separation of the placenta from the maternal blood supply or even the death of the mother, would all cause in utero the hypoxic stimulus for gasping analogous to the plastic bag after birth.

If the infant monkey had not been resuscitated, the complete asphyxia would have led to death. However, the studies were not done to define the conditions producing death, but to discover the conditions that produce cerebral palsy. This complete asphyxia model had a serious flaw. The experiments produced brain lesions confined to nuclei in the brain, and this was not usual anatomic finding of extensive white and gray matter necrosis observed in children with cerebral palsy.
As a result, Dr. Ron Myer was funded to continue the study of fetal asphyxia in a free-range monkey colony on a small island near Puerto Rico. His initial studies reprised those of Dr. Dawes and others. A major epiphany occurred following a chance variation in the experimental protocol7. A mother monkey became agitated and demonstrated an acidemia. The fetus had a moderate acidemia at birth. The researchers proceeded with the experiment in the usual manner, but they had a very different result. The infant monkey had a prolonged post-asphyxial neurological deficit including seizures; he had flame shape retinal hemorrhages; and he eventually developed symptoms consistent with cerebral palsy. He was sacrificed at 6 months of age and the brain showed cortical atrophy and gliosis, and basal ganglia gliosis similar to that seen in humans with cerebral palsy. Dr. Myer reasoned that it was the prenatal acidosis that made the difference.
Further experiments created intrauterine hypoxia, and hence fetal acidosis, by various manipulations including duplicating maternal stress with epinephrine, using heavy maternal halothane anesthesia, and compressing the maternal aorta to slow uterine circulation8. The experiments further demonstrated that a plastic bag over the newborn was not required. The fetal hypoxia itself led to the terminal cardiovascular collapse of the fetal monkey9. This collapse was likely due to a positive feedback loop of progressive acidosis leading to decreasing cardiac contractility leading to worsening hypoxia. This collapse presented a problem for the experiments in that the fetal monkey often died before the experimenters could perform resuscitation.
For the purpose of understanding a mechanism of stillbirth, the experiments demonstrated that partial asphyxia alone would eventually lead to hypoxia sufficient to stimulate gasping, and to cause fetal death. In Myer’s experiments, the degree of hypoxia was often severe (but still less than complete) leading to death or brain lesions in 20 to 30 minutes. Some clinical cases provide evidence of lethal partial asphyxia of much longer duration. These cases need to be distinguished from chronic fetal hypoxia often from decreased utero-placental blood flow or compromised placental anatomy. A reasonable distinction is that partial asphyxia causes an unrelenting, increase in fetal acidosis leading to cardiovascular collapse and death. Chronic hypoxia leads to placental and fetal adaptations that increases fetal oxygenation and sustains fetal life.
A refinement on the definition of partial asphyxia was made in ovine studies with a doughnut like occluder wrapped around the umbilical cord with a timer set to regularly occlude the blood flow for a fixed duration at fixed intervals. This intermittent asphyxia within limits can result in fetal lamb acidosis and brain injury equivalent to partial asphyxia10.
Asphyxia can be interrupted by resuscitation, either by medical intervention, or by natural changes in the conditions causing the hypoxia. Multiple episodes of non-lethal asphyxia could occur in utero if there was a reversible condition. Lesions of hypoxic ischemic brain injury found in stillborn infants is direct evidence of this recurrent fetal asphyxia11. In recurrent asphyxia, the fetus would resolve the hypoxia/acidemia between episodes of asphyxia rather than having a sustained progression of acidosis.
A reasonable conclusion is that the intrathoracic petechiae and findings of acute heart failure are caused by fetal asphyxia, either complete, partial or intermittent. This conclusion can be tested by identifying the same lesions in a known cause of compete or partial asphyxia, namely stillbirth from premature placental separation. Clinical placental abruption is one consequence of a large placental separation, but some separations may be clinically silent. Almost all separations develop a hematoma between the placenta and uterus which depresses the maternal surface. The placenta above the separation is devitalized showing changes similar to the stages of placental infarction. The pathologist can use these features to estimate the loss of functioning placental volume. If the separation is complete, the fetus suffers acute total asphyxia. Smaller separations are equivalent to partial asphyxia. An example of partial asphyxia is provided by the case of a gravid, near-term woman who suffered a subdural hematoma from an auto accident. She had normal fetal heart tones at the time of hospital transfer at 4 hours after the accident. In the receiving hospital, the heart tones were found to be absent 4 hours later. A stillborn infant was delivered 4 hours after loss of fetal heart tones. The placenta demonstrated a 40% loss of viable placental tissue. The deceleration forces in an auto accident are believed to be a cause of premature placental separation12. A reasonable interpretation of the case is that a 40% placental separation at the time of the accident caused partial asphyxia that caused fetal death somewhere between 4 to 8 hours later.
A retrospective study of stillborn autopsies attributed to placenta separation ranked the placentas by the extent of separation and correlated that extent with microscopic petechiae and vascular congestion13. The petechiae were present in all cases with up to 75% separation (Fig 3). Smaller separations did not always show intrathoracic petechiae. A reasonable conclusion is that petechiae were associated with severe intrauterine asphyxia. A possible explanation for the absence of intrathoracic petechiae with lesser degrees of separation is that with less hypoxia, the slower development of brain acidosis anesthetized the brain blocking the signal to start breathing.

Since petechiae confined to the intrathoracic surfaces are found in many stillborn infants of unknown cause, a reasonable inference is that those stillborn infants also suffered acute severe asphyxia. In stillborn infants showing only evidence of acute congestive heart failure, asphyxia is not excluded, but should may correspond to a mechanism of less severe hypoxia than in those infants with petechiae.
The pathological evidence has led to the hypothesis that some stillbirth without a cause of death are due to asphyxia. This hypothesis does not indicate the mechanism of the asphyxia. Without a visible placental lesion or a convincing maternal history of asphyxia, suspicion falls on the umbilical cord. Many have noted that the umbilical cord is the lifeline of the fetus, much like a deep sea diver’s umbilical line. The experimental studies of umbilical cord induced fetal asphyxia provide no insight in that they relied on manufactured occluders, not on a natural mechanism. A meta-analysis of published reports of umbilical cord and stillbirth found very few acceptable papers for analysis14. Of the lesions considered, only two correlations with stillbirth were significant, true knots of the cord, and multiple nuchal cord wrappings. Both conditions are rare, and both require deeper examination as to how they result in fetal death which will be explained later.
There is some indirect evidence that decreased umbilical blood flow may be involved in unexplained stillbirth. Parast and colleagues found a correlation of unexplained stillbirth with thrombi in chorionic blood vessels and with fetal vascular malperfusion lesions in the villi (Fig 4)15. Thrombi require flowing blood to form and by definition are premortem. A reasonable argument going back to Virchow’s triad for thrombosis, namely, stasis, vascular injury, and thrombophilia, is that decreased umbilical blood flow led to the thrombi in the fetal circulation in the placenta similar to that of deep vein thrombosis in the legs. The villous malperfusion lesions are due to stasis of blood in vessels kept viable by the surrounding maternal circulation. These may form pre or postmortem. Some are acute, villous karyorhexisis, but others require time to develop such as cell proliferation or stromal changes of avascular villi. A subsequent study associated similar lesions in a variety of umbilical cord abnormalities, that along with the focal distribution, supported that the hypothesis that lesions are the result of umbilical blood flow compromise16. A surprising conclusion from these observations is the implication that the conditions of fetal asphyxia had been present for days or even weeks before death based on estimates of the development of similar lesions with postmortem retention17.

The left shows a dilated hemoglobin stained vessel with arrows pointing to discrete calcific thrombi. The right shows a stem vessel with intimal proliferation and fragmented red cells, the results of vascular stasis, a fetal vascular malperfusion lesion.
Another possible insight into the fetal vascular lesions in stillborn placentas comes from an observation made during attempts at fetal lamb heart surgery on by-pass. The researchers found that they could not restart the fetal placental circulation after surgery because of a severe increase in vascular resistance in the placental bed18. They attributed this to the lack of blood flow during the surgery triggering an intrinsic vasopressin induced constriction of the placental vessels that could not be reversed. Perhaps this is not surprising as the umbilical arteries respond with permanent constriction after delivery of the infant. Hypothetically, some regions of the placental circulation may be more prone to constrict in response to a general decrease in umbilical blood flow. The role of vascular contractility in response to decreased umbilical blood flow may play a role in exacerbating fetal asphyxia.
Aside from these secondary placental lesions, postulating decreased umbilical blood flow as the mechanism of fetal asphyxia is frustrated by the simple observation that in most cases of unexplained stillbirth, there is no anatomic lesion of the umbilical cord. One explanation is that the anatomic occlusion does not harm the cord per se. To test this hypothesis, I step sectioned several cords that had been used with occluders in sheep studies, generously sent to me by Dr. Laura Bennett. The location of the occluder on the cord was unknown, so in that sense I was blinded as I would be in a stillborn infant. I could not find a lesion produced by the occluder grossly or microscopically. I have to accept that the lesion can be undetectable despite severe compression. As a pathologist, I occasionally see in random umbilical cord microscopic samples a focus of necrosis and even calcification in an umbilical vein from liveborn deliveries without a history of asphyxia, but this could be a marker of transient injury to the cord.
Compression in occult prolapse of the umbilical cord is plausible cause of compromise of umbilical blood flow even prior to labor if the cord is wedged between a fetal part and the pelvis. In some cases of cord prolapse in stillbirth, the compression is evident by color differences in the cord (Fig 5).

However, there may be another simple overlooked mechanism of cord compromise. As any gardener will tell you, a sudden kink in a garden hose can stop the flow. Looking at the kinks closely, they appear to be from torsion on the hose causing a loop to form in which the lumen collapses. Does this occur with the umbilical cord? As a summer project with a medical student, we tried to answer this question empirically by perfusing umbilical cords in vivo and wrapping them in various way around a baby doll (which eventually was replaced by segments of PVC pipe) (Fig 6).The student noted that twisting the short leftover end of the cord after wrapping it around a pipe was the most effective way to stop flow. The shorter the segment, the less rotation was required to stop the flow19 (Fig 7). Since the study was not physiologic, the in vivo results might have different magnitudes, but the principle should hold. At first, I thought the result was trivial, but in discussing with obstetricians, it became clear that this was an overlooked clue to the mechanism of umbilical vascular occlusion. If in utero, a cord becomes wrapped around the fetus near the placenta or with multiple wraps, leaving a short portion of free cord between the wrapped end and the placental insertion, this segment would become a functionally short cord (Fig 8,9). As in our in vitro experiments, the cord blood flow would become more susceptible to compromise by torsion. A functionally short segment of cord would put more torque on the cord insertion with fetal turning. True knots of the cord form as a fetus “swims” through a loop of cord, but until the knot becomes tightened, it may not produce interference to blood flow. Fetal wrapping that shortens the free cord length could apply tension to tighten the knot.



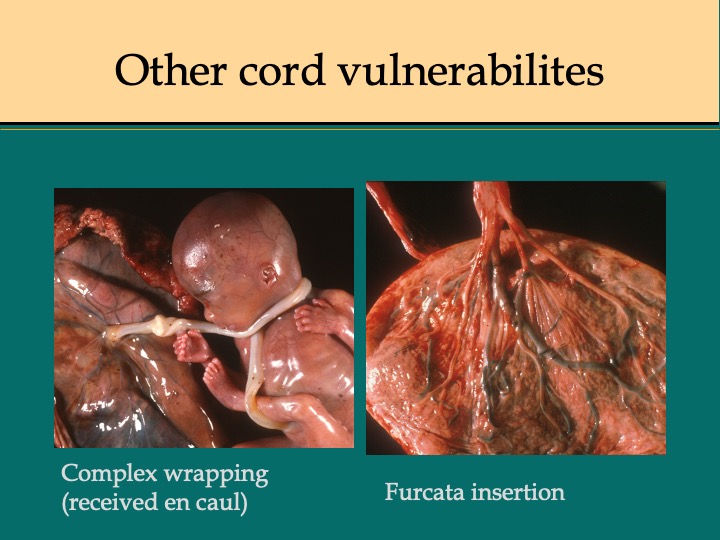
These observations and thought experiments lead to the hypothesis that a functionally short cord from fetal wrapping may compromise umbilical blood flow. To prove this hypothesis would require a detailed visualization of the cord anatomy in situ before delivery. At a meeting of the parents-of-stillbirth group, Star Legacy, Dr. Jason Collins proposed that this could be done with an MRI. Proposing such a study to a mother who has just discovered that she lost baby would not be easy, but the benefit to others would be large if a cord abnormality, such as short functional cord, proved to be a key factor in stillbirth. The same MRI study could also identify occult prolapse or other forms of compromised umbilical cord.
Dr. Collins added another important corollary to the basic hypothesis, namely that lethal asphyxia may occur from a convergence of events that include compromise of the umbilical cord. He had interviewed a large number of mothers of stillbirth over the internet. He found a disproportionate number had gone to bed with normal fetal movement and awoken with loss of movement. Sleeping could compound an already compromised umbilical blood flow by decreasing oxygenation or increasing maternal acidosis through sleep apnea, supine sleeping with pressure on the aorta, or fasting ketosis at night. The idea of a necessary convergence of insults might explain recurrent fetal asphyxia and natural resuscitation, for example by changing sleep position, that would explain fetal vascular lesions of weeks duration. The multi-factorial model could explain how risk factors impact individual infants. For example, sleeping with pressure on the aorta may be more important in obesity, or ketoacidosis may be a more significant problem in a diabetic mother. Likewise, genetic differences in the mother or fetus could affect fetal response to hypoxia or acidosis. The functional reserve of the placenta for oxygen transfer to the fetus may be yet another factor in susceptibility to hypoxia.
As presented at the 2019 Star Legacy meeting, recent programs to reduce stillbirth, including encouraging mothers to sleep on their side, show some very preliminary success These programs also encourage mothers to learn their baby’s movement pattern and report any change to the caregivers. This appears to be a more sensitive marker of fetal distress than kick counts. Such information may also be valuable in retrospect as a perceived increase in activity could be from the onset asphyxia and fetal gasping. Such observations may be of value in explaining anatomic evidence of recurrent asphyxia in stillbirths.
This model of concurrent factors expands the concept of causation in stillbirth. Umbilical blood flow compromise has a high probability of being one frequent factor in causing fetal death. The model of partial asphyxia and progressive fetal acidosis as sufficient to cause fetal death, provides a framework for tying other risk factors to the immediate cause of death.
A study of stillbirth-of-unknown-cause should reasonably start with identifying umbilical cord configurations in situ as the cornerstone to correlate with the other anatomical and clinical findings. If the hypothesis of a key role for compromised umbilical blood flow is true, then an MRI will uncover a pattern such as functional short cord from wrapping or unsuspected occult cord prolapse.
A second part of the study would seek to increase the details obtained in clinical and laboratory studies in the mother to uncover contributory factors in the model of a multi-factorial cause of death. Questions would need to be developed for the parents about sleep patterns, fetal movement, diet patterns, minor illness or other problems and events not yet anticipated that were occurring around the time of fetal death. The laboratory markers need to be sought to elucidate perimortem events, such as maternal urine ketones. A protocol for placental examination and autopsy directed to the umbilical cord, fetal vascular circulation and placental oxygen reserve would need to be developed.
The reason this in an invitation and not a proposal is that only by brainstorming sessions with people with a broad range of knowledge can the best proposal be developed. Participants need to include obstetrical care givers, radiologists, pathologists, behavioral scientists and caregivers, basic scientists, and parents of stillborn infants. A pilot protocol would be developed and then review of each case would be made with the intent of improving the protocol by iteration. The goal is that this study on stillbirth will improve care for the parents, and lead to improvements in intervention to prevent stillbirth.
Even though the methods in this study would be prospective, the diagnosis of a stillbirth of unknown cause is made after the autopsy is complete. My estimate of the incidence of stillbirth without a known cause, from my database of autopsies I have performed, is approximately 20%. Published estimates vary widely. Even with stillbirth occurring in one in 160 deliveries, a single institution would have only a limited number of cases with consent per year. A pilot study would provide a better estimate of cases. A successful pilot study would also open the door to multi-institutional studies that would provide adequate numbers of study participants.
If intrauterine cord configurations, such as functionally short cord, prove to have significant associations with stillbirth, this observation could provide the basis for funding experimental models to that could cause and relieve umbilical blood flow compromise. Further research into anatomic cord mechanisms of umbilical blood flow compromise should also contribute to understanding and preventing hypoxic ischemic brain injury in live born infants.
References:
1. Reddy UM. Stillbirth. Foreword. Clin Obstet Gynecol 2010;53:586-7.
2. Dudley DJ, Goldenberg R, Conway D, et al. A new system for determining the causes of stillbirth. Obstet Gynecol 2010;116:254-60.
3. Pinar H, Koch MA, Hawkins H, et al. The Stillbirth Collaborative Research Network (SCRN) placental and umbilical cord examination protocol. Am J Perinatol 2011;28:781-92.
4. Parker CB, Hogue CJ, Koch MA, et al. Stillbirth Collaborative Research Network: design, methods and recruitment experience. Paediatr Perinat Epidemiol 2011;25:425-35.
5. Harrison MS, Thorsten VR, Dudley DJ, et al. Stillbirth, Inflammatory Markers, and Obesity: Results from the Stillbirth Collaborative Research Network. Am J Perinatol 2018;35:1071-8.
6. Dawes G. Foetal and Neonatal Physiology. Chicago, IL: Year Book Medical Publishers, Inc.; 1968.
7. Myers RE. Atrophic cortical sclerosis associated with status marmoratus in a perinatally damaged monkey. Neurology 1969;19:1177-88.
8. Myers RE. Two patterns of perinatal brain damage and their conditions of occurrence. Am J Obstet Gynecol 1972;112:246-76.
9. Myers RE, de Courten-Myers GM, Wagner KR. Effect of Hypoxia on Fetal Brain. In: Beard R, Nathanielsz PW, eds. Fetal Physiology and Medicine, Second Revised Edition. New York: Marcel Dekker, Inc.; 1984:419-58.
10. De Haan HH, Gunn AJ, Williams CE, Gluckman PD. Brief repeated umbilical cord occlusions cause sustained cytotoxic cerebral edema and focal infarcts in near-term fetal lambs. Pediatr Res 1997;41:96-104.
11. Grafe MR, Kinney HC. Neuropathology associated with stillbirth. Semin Perinatol 2002;26:83-8.
12. Stafford PA, Biddinger PW, Zumwalt RE. Lethal intrauterine fetal trauma. Am J Obstet Gynecol 1988;159:485-9.
13. Bendon RW. Review of autopsies of stillborn infants with retroplacental hematoma or hemorrhage. Pediatr Dev Pathol 2011;14:10-5.
14. Hayes DJL, Warland J, Parast MM, et al. Umbilical cord characteristics and their association with adverse pregnancy outcomes: A systematic review and meta-analysis. PLoS One 2020;15:e0239630.
15. Parast MM, Crum CP, Boyd TK. Placental histologic criteria for umbilical blood flow restriction in unexplained stillbirth. Hum Pathol 2008;39:948-53.
16. Tantbirojn P, Saleemuddin A, Sirois K, et al. Gross abnormalities of the umbilical cord: related placental histology and clinical significance. Placenta 2009;30:1083-8.
17. Genest DR. Estimating the time of death in stillborn fetuses: II Histologic evaluation of the placenta: a study of 71 stillborns. Obstet Gynecol 1992;80:585-92.
18. Lam CT, Sharma S, Baker RS, et al. Fetal stress response to fetal cardiac surgery. Ann Thorac Surg 2008;85:1719-27.
19. Bendon RW, Brown SP, Ross MG. In vitro umbilical cord wrapping and torsion: possible cause of umbilical blood flow occlusion. J Matern Fetal Neonatal Med 2014;27:1462-4.
