In the page on umbilical cord accident I discussed the ways in which blood flow through the umbilical cord could be occluded by compression, either directly or through torsion, or even through arterial wall contraction. Another way in which fetal blood flow can be stopped is clotting of blood within the umbilical and placental vessels, pathologically defined as thrombosis or thrombus formation. Distal to such a thrombus, the lack of blood flow produces distinctive lesions that puzzled pathologists in the past, but currently are considered features of fetal vascular malperfusion (FVM). If the fetal circulation demonstrates a thrombus, the relationship to the distal lesions is evident. However, if the FVM lesions are present and a thrombus is not detected, then other chronic causes of vascular occlusion, fetal death, or failure to find the culprit thrombus are all possible explanations of the findings. In the placental circulation, occlusion of either a chorionic vein or artery will result in a villus volume of the placenta without blood flow, as the stem vessels of the placental architecture in general do not anastomose with each other. After considering the pathological lesions I will discuss the causes of thrombosis in fetal vessels, as well as the consequences. This page will end with a discussion of the evidence that even sparse fetal thrombotic vasculopathy/vascular malperfusion can be a marker of umbilical cord compression as a cause of fetal death.
Thrombi in the umbilical cord and in the fetal vessels in the placenta
As every student of pathology learns, a thrombus forms in flowing blood as the consequence of activation of clotting that starts on the inner lining of the vessel. Waves of activation of thrombin formation from its precursor thrombinogen and platelet activation result in a laminated thrombus that contains relatively dense fibrin. Fibrin is the protein that stops the flow of blood and normally is controlled by an active balance of processes that cause fibrin to form (coagulation) from the precursor in blood (fibrinogen) against those processes that digest fibrin (fibrinolysis). In the slow blood flow of a vein the formation of a thrombus shows telltale lamination (layers) that can be seen under the microscope. Blood trapped in a vessel can coagulate to form a fibrin clot but it is less dense than a thrombus, and still has an elastic quality that is lost in a thrombus. The ultimate proof that clotted blood is a thrombus formed during life is that the thrombus begins to organize, a term of art in pathology. Organization is the ingrowth from the vessel wall of fibroblasts and new capillary blood vessels that will in time attempt to form a new blood flow channel or channels in the vessel, and will reduce the fibrin to a collagen scar. The former occurs from the linking of the new vessels and the latter is the result of fibroblasts and macrophages remodeling the thrombus into scar. This process occurs over time and may not be present in a recent thrombus. The pathologist must use judgment based on the histology to determine whether a placental vessel shows a thrombus or just a postmortem/post delivery clot.
An occlusive thrombus in the umbilical vein would reduce or end oxygenated blood flow to the fetus. I have found such thrombi in stillborn infants but as we will see, there are no criteria to determine if the thrombus was a consequence of umbilical cord compression or the primary event that caused fetal death (Fig 1, 2).
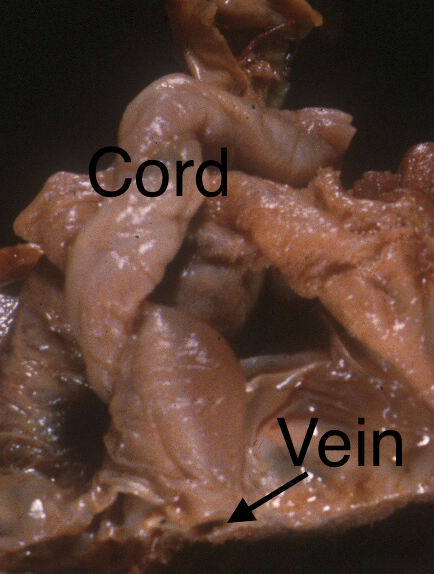
Fig 1a: The cord insertion has been cut and the dilated vein demonstrated thrombus at the arrow. This stillborn infant had meconium staining and aspiration.

Fig 1b: This photograph of the sliced placenta on its side also shows the section of thrombosed umbilical vein (arrow).

Fig 2a: This is a cross section of the umbilical vein showing a partially occlusive thrombus (marked). This stillborn infant had otherwise unexplained fetal hydrops. (H&E 4x)
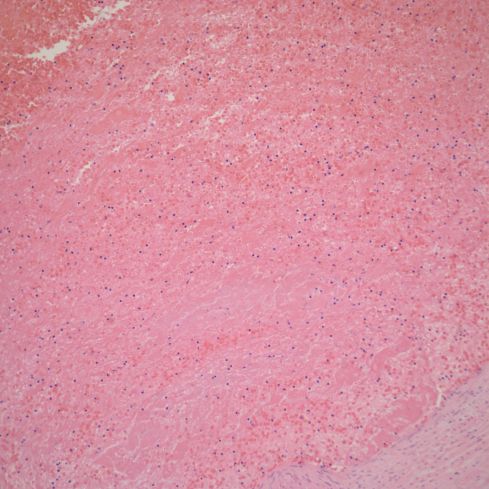
Fig 2b: A higher power magnification of the thrombus showing subtle lamination of fibrin in the thrombus.
As presented in the section on umbilical vein hematoma and aneurysm, non-occlusive thrombi can form in the wall of an aneurysm, as well as be found on the umbilical vein as an incidental finding (Fig 3). Thrombi that appear to be occlusive in the umbilical vein are rare.

Fig 3: This is a mural thrombus in the umbilical vein (*’s)
In contrast, occlusive thrombi in an umbilical artery are relatively common. Of course, an occlusive arterial thrombus in a fetus with a single umbilical artery would reduce blood flow to the placenta and cause fetal hypoxia and death. I have never seen a case of this occurring. An occlusive thrombus in one of two umbilical arteries in my experience is often an incidental finding in a normal pregnancy and there are case reports of relatively unaffected infants with such thrombi[1-3]. In the human, almost all placentas have an anastomosis of Hyrtl near the placental insertion that allows one artery to perfuse the whole placenta if the other is blocked[4]. In the sheep, which has separate cotelydons without arterial anastomoses, ligation of one umbilical artery results in loss of fetal perfusion to half the placental mass[5]. Rarely, but analogously, the same loss of placental perfusion occurs in humans if the placenta has no Hyrtl anastomosis or if the thrombus occludes the anastomosis[6, 7].
Some published series of umbilical artery thrombosis report a high incidence of complications including stillbirth and growth restriction[6, 8]. One possible reason that this differs from my experience is that the reports are from placentas examined in pathology because they had an obstetrical complication, and hence are a biased sample. Another reason may be that I often make the diagnosis by inferring a thrombus by the sometimes subtle findings in a necrotic artery. These changes could be overlooked.
The histologic changes that I look for are the difference between one viable contracted, umbilical artery and another necrotic dilated artery. From this difference, I infer a thrombus somewhere in one umbilical artery. The reasoning is such. A thrombus at any level of the umbilical artery would stop forward flow in the entire artery and back flow from the anastomosis of Hyrtl would fill the artery downstream to the occluding thrombus. Without blood flow, the arterial wall dies and remains dilated with blood. Not only does the artery die, but so do the trapped red blood cells. Hemoglobin diffuses from the dead red cells into Wharton’s jelly creating a red cord often with a “candy-stripe” helix that follows the artery (Fig 4).

Fig 4: The umbilical cord has a helix of red stained Wharton’s jelly similar to that found in fetal postmortem retention, but it follows only one vessel and the surface of the placenta demonstrates normal appearing vessels without the similar red brown staining seen with fetal death.
Fetal death creates a uniformly red cord since hemoglobin diffuses from both arteries and the vein (Fig 5).

Fig 5: This is a red umbilical cord attached to a stillborn infant with 48 hours of postmortem intrauterine retention.
The important diagnostic clue is that the cord has a red discoloration, but the infant is liveborn. Under the microscope the artery wall shows coagulation necrosis and the lumen may show “ghosted” (no hemoglobin) red cells and some fibrin (Fig 6).
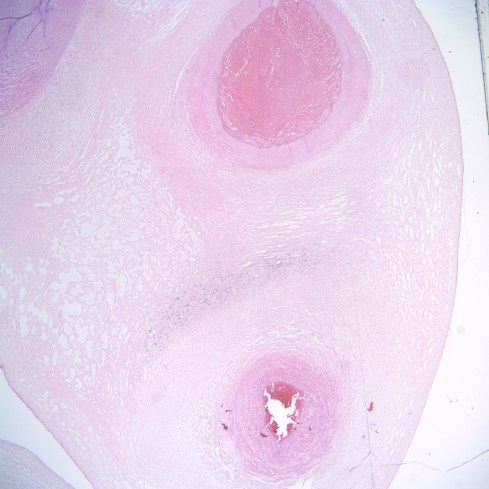
Fig 6a: This low power cross section of the umbilical cord demonstrates a constricted, viable artery in the upper left corner and a vein in the bottom. In the right corner is a dilated necrotic artery. Between the vein and the necrotic artery is a crescent of blue due to inflammatory cells that have migrated from the vein to the necrotic artery. Inflammatory cells would be expected to migrate to an area of necrosis. However, the unusual geometry of that migration is due to the lack of capillaries, forcing the cells to leave the large umbilical vein more of less uniformly. (H&E, 2x)

Fig 6b: This higher power of the necrotic artery demonstrates thrombus as strands of dense fibrin along the inner surface and in the lumen. Fragments of red cells have been forced into the intima of the artery. Some of the red cells still appear bright red while others are colored only by the pink eosin used to stain the section. (H&E,20x)
A laminated thrombus is seldom found in our random cord sections, likely because the mechanism is unlike that of a low-pressure venous thrombus. Rarely there is a mural arterial thrombus, and this may show organization showing it occurred some time prior to complete occlusion. This organization stops if the arterial flow stops because the entire structure becomes hypoxic and then necrotic (Fig 7).

Fig 7a: This low power cross section of an umbilical cord shows a necrotic dilated artery in the right upper corner. (H&E, 2x)

Fig 7b: This higher power of the same artery demonstrates organization with early fibroblast ingrowth and neovascular channels. These features have also become necrotic. (H&E, 10x)
In some cases a very diminutive necrotic artery is found suggestive of a thrombus early in gestation or even a developmental hypoplasia of the artery (Fig 8).

Fig 8a: This is a low power cross section of the umbilical cord with a hypoplastic artery in the upper right corner. (H&E, 2x)

Fig 8b: This higher power of the same artery shows necrosis in the muscularis (the fibers are pinker and show loss of circumferential orientation) and the lumen shows stasis and fibrin. (H&E, 20x)
Arterial thrombi may not only be clinically silent, but also show no other thrombi or secondary evidence of thrombi in the placenta.
The fetal chorionic vessels are end vessels once they enter the placental villi and a thrombus in such a vessel will result in a loss of placental function including respiratory gas exchange in the villi supplied by that vessel. That loss of function is implied by the term “end vessel” meaning there is no other way for blood to reach a functioning part of an organ except by the given end artery. A thrombotic embolus occurs when a piece of a thrombus is shed into the circulation and gets stuck further downstream in the circulation. An embolus from a thrombus in the umbilical artery would enter an end vessel of a placental villus and fetal blood flow downstream would stop. In my experience, evidence of such emboli with umbilical artery thrombi is rare. I have seen one case in which there was an umbilical artery thrombus, multiple villous vessel thrombi that may have been embolic, and fetal distress during labor that likely was due to the loss of perfused placental tissue [9](Fig 9).

Fig 9: The umbilical cord demonstrates a red candy stripe pattern from hemoglobin diffusion from a single occluded artery. The arrow shows one of the umbilical arteries that demonstrated occlusive emboli.
The pre-delivery ultrasound examination demonstrated a thrombus in the fetal aorta. The placental emboli could have come from the aorta, not the thrombosed umbilical artery.
Umbilical artery thrombus may have some of the same etiologies discussed later for downstream thrombi and thrombi in the umbilical vein. However, there could be a unique mechanism applicable to some umbilical arterial thrombi. Perhaps some of the occluded umbilical arteries that I have seen are not caused by thrombus, but by prolonged contraction of the artery. This speculation could be proven if complete sampling of the cord fails to find a thrombus in placentas with umbilical artery necrosis.
Thrombi in the placental circulation
To understand thrombi in the placental circulation, one needs to understand the placental circulation. In a reversal of their usual roles in the body, the umbilical arteries carry unoxygenated blood to the placenta. These two arteries arise from the iliac arteries that in the adult carry oxygenated blood to the legs and pelvis. They do not carry oxygenated blood in the fetus because of the unique fetal circulation: The arterial blood becomes oxygenated in placental capillaries. This blood exits through the placental veins to the umbilical vein, then through the ductus venosus into the liver and then into the inferior vena cava. The opening of the inferior vena cava is directly opposite an opening between the atria of the heart, the foramen ovale. The oxygenated blood enters through this foramen into the left atrium. The left atrium after birth will receive oxygenated blood from the lungs, but in utero the high pulmonary arterial resistance reduces pulmonary blood flow to only a small amount. Most of the blood entering the left atrium is from the oxygenated umbilical vein. This oxygenated blood enters the left ventricle and is pumped out the aorta. Thus, the fetal circulation maximizes oxygenated blood going to the heart via the coronary arteries and to the brain via the carotid arteries and vertebral arteries. On the right side of the heart, unoxygenated blood from the superior vena cava is pumped out the pulmonary artery, and most of this blood is shunted through the ductus arteriosus to the aorta just below the subclavian arteries. After birth, the ductus and foramen ovale will close and pulmonary vascular flow will increase as respiration decreases pulmonary arterial resistance. Until birth, the fetal circulation causes the inverted arterial and venous circulation of the placenta.
The umbilical cord, as we saw in the illustrations of modes of placental insertion in the last chapter, rapidly enters a branching pattern of vessels on the surface of the placenta. The surface vessels are often referred to as chorionic vessels, although all vessels in the placenta are chorionic, i.e. placental. All placental vessels fail to show the typical anatomic differences under the microscope that distinguish a vein from an artery. The identity of the vessel as vein or artery can be determined by tracing its origin from the cord arteries or vein. More simply, the vessels can be identified by the curious observation that the arteries always cross over the veins (Fig 10).
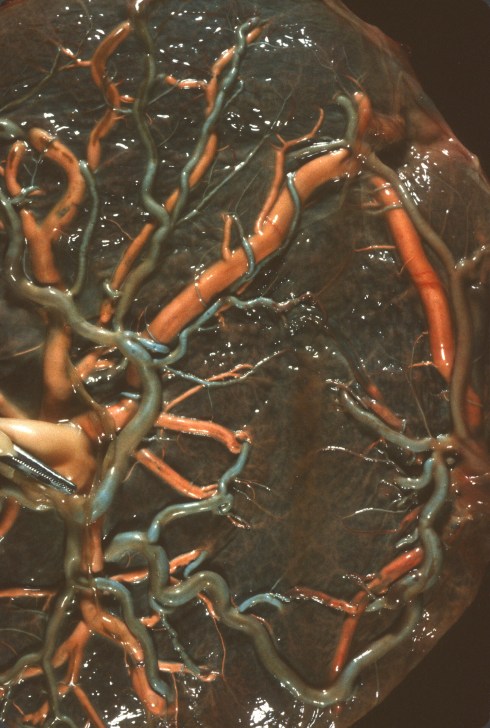
Fig 10: In this twin placenta, the umbilical artery was injected with blue dyed barium and the vein with white barium. Everywhere on the fetal surface of the placenta, the blue arteries are overriding the veins.
The chorionic vessels then branch “downward” into the villous “tree” of the placenta. A stem artery enters the trunk of a villus and its branches follow the villous branching into capillaries in the terminal villi and these in turn are connected to the branching veins until they enter a stem vein at the same trunk of the primary villus. The villi dangle in the maternal blood and there are no connections between them. If flow to a stem artery or vein is occluded then fetal blood flow to and from the villus is stopped, as noted above these are end arteries, and also in effect are end veins. Maternal blood flow continues to nourish the affected villus. This is a simplified description of the fetal placental circulation, but it is sufficient to understand the next part of the discussion.
In an early study of villous arterial thrombi in the placenta published by Dr. Harold Fox, he noted that a single fetal stem thrombus was an incidental finding, but that in three stillbirths without prolonged in utero retention, there were multiple arterial thrombi with organization of the thrombi (in the pathology use of the term) that was more advanced than the period of retention of the fetus after fetal death[10]. This 1966 paper in the very first paragraph succinctly describes the pathological appearance of thrombosis of a fetal vessel in the placenta. The paper also directly and indirectly noted some of the difficulties that would continue to puzzle pathologists in the coming years. First, he could not find a pathological cause of the thrombi. Second, he identified fetal vascular changes in the placental of stillborn infants with prolonged postmortem intrauterine retention that resembled those in the placenta with isolated thrombi. He presumed, but did not prove, that the thrombi were arterial because of the loss of circulation in a pattern of the distal stem villous. He argued that the organized thrombi in the placentas of the stillbirths were evidence that the thrombi had preceded the fetal death and could therefore in themselves not have been the cause of death. However, he noted in these cases that the placenta distal to the thrombus was non-functional and this loss of reserve function could have contributed to fetal death. The prescience of his observations, as well as the error of assuming that the thrombi were all arterial, will become evident in the following discussion.
The thrombi that Dr. Fox was describing were occlusive, that is, they blocked all of the blood flow through the vessel (Fig 11).
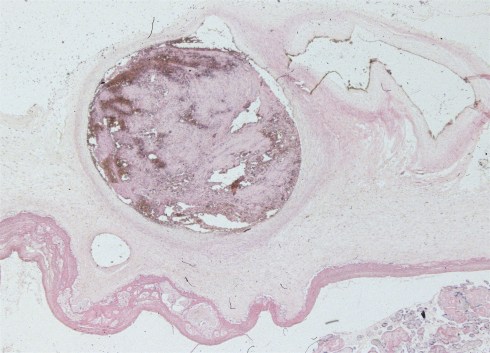
Fig 11a: This image is through the placental surface and shows a loose occlusive thrombus in a chorionic surface vessel. (H&E, 2x)
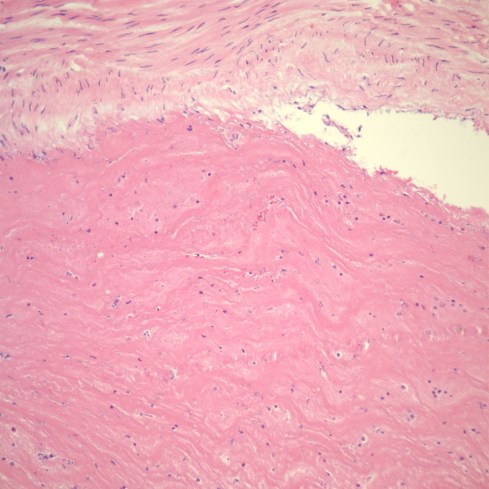
Fig 11b: This image shows an occlusive thrombus with dense lamination of pink fibrin strands. (H&E, 20x)

Fig 11c: This occlusive thrombus shows organization with small new capillaries, fibroblasts, and some inflammatory cells at the intimal edge of the thrombus (arrow). (H&E, 20x)
Thrombi may hug the vessel wall and not block blood flow (Fig 12).

Fig 12a: Partially occlusive thrombus with lamination of blue cellular layers and fibrin layers. (H&E, 2x)

Fig 12b: Mural thrombus of dense fibrin in a surface vessel.(*) (H&E, 4x)
In 1973 Dr. DeSa published a paper “Intimal Cushions in Foetal Placental Veins” which was primarily concerned with observations on thrombi in veins on the placental surface[11]. The lesions described, including some in stem vessels, covered a variety of lesions. Those described as major intimal cushions likely were organizing thrombi. The end result of organization may be a small cellular scar protruding from the vascular intima (Fig 13)
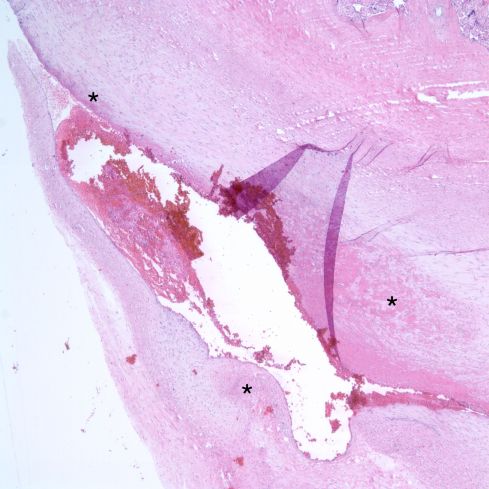
Fig 13a: Mural fibrin thrombus showing various degrees of organization of fibrin (asterisks) in a oblique section of a surface chorionic vessel (H&E 4x)

Fig 13b: Higher power of mural fibrin showing ingrowth of pale scar, fibroblasts and macrophages between strands of fibrin (H&E, 20x)

Fig 13c: This longitudinal section of a surface chorionic vessel shows a well-formed intimal cushion presumed to be from an organized mural thrombus (arrow). This placenta showed occlusive thrombi in other sections. (H&E, 2x)
A paper by Dr. Jean Scott in 1983 entitled “Fibrinous vasculosis on the human placenta” described vascular lesions in the stem villi[12]. This pioneering study added a layer of terminology of organized thrombi in the placental vessels that is no longer used. However, I want to discuss one observation from that paper: “In most instances the artery was contracted and empty, but the vein, which tended to have more oedema in its wall, was seen on longitudinal section to have irregular areas of contraction and dilatation. As a result of this, oedematous subendothelial cushions formed near contracted areas {figure 3}. Where the lumen narrowed platelet fibrin thrombi were frequently found {figure 4}”. The photographs look like the intimal cushions that can be found in many placentas from uncomplicated pregnancies without other evidence of fetal thrombi. The term cushion here is not referring to the loose, circumferential intimal connective tissue in the umbilical arteries, but to a raised area of intimal hyperplasia often with a fibrin cap and sometimes calcification. In chance longitudinal sections of the villi, the cushion appears to be at a branch point (Fig 14).

Fig 14a: This is a longitudinal section through a stem villous branch that shows a vessel with an intimal fibrous cushion topped with pink fibrin. (H&E, 2x)

Fig 14b: This is a higher power magnification of the above intimal cushion.
While some cushions are likely organized mural thrombi that develop from a pathological process, others might plausibly be due to turbulence in the placental circulation. These latter cushions may not have the same pathological implications that we will discuss for other placental vascular thrombi. Further study would need to identify cushions at branch points and then correlate this specific lesion with the clinical outcome in a series of unselected placentas and that would be a difficult task to accomplish.
Small fibrin thrombi can also be found in the smallest vessels of placental circulation, the villous capillaries. The thrombi are not laminated and do not usually show organization. The end result is likely just the loss of the capillary with its thin endothelial wall.
The effects of lost blood flow on the placental anatomy
Before considering the clinical meaning of placental thrombi, the lesions that these thrombi can cause in the placenta needs to be presented. A thrombus occurring at just one point in the fetal circulation can have consequences through the whole end circulation of the villi. The consequences are similar whether the thrombotic blockage is in the artery or vein, as blood will stop flowing in the villus. The villi can be seen as trees inverted into the maternal blood stream. This area of secondary effect is usually much larger than that of the thrombus, making its identification on a microscope slide more likely than that of the thrombus itself. Thus, the diagnosis of fetal vascular malperfusion often hinge on identifying these secondary lesions.
An occlusive thrombus in a stem villous vessel will stop blood flow yet there will remain a persistent blood pressure on both sides of the thrombus that will keep the vessels distended. There is backpressure on the venous side, and forward arterial pressure on the forward arterial side. The stagnation of blood results in ischemic injury to the endothelial cells lining the vessels and this is manifested under the microscope by red cells forcing their way past this intimal lining (diapedesis). The maternal circulation between the villi keeps the villus alive after its fetal blood flow has stopped. These events produce a distinctive pathological lesion that was a topic of intense study by Dr. Sander at the Michigan Placental Registry[13-20]. Dr. Sander first published about this lesion in 1980 and the first sentence of that paper begins: “ A previously undescribed pathologic alteration has been observed to date in 32 specimens…[13]” That paper showed striking images of the “intraluminal fragmentation of erythrocytes with diapedesis of intact and fragmented RBCs through vessel walls”. The lesion was named hemorrhagic endovasculitis (HEV) and considered evidence of a microangiopathic process, perhaps due to viral infection (Fig 15).

Fig 15: This is a typical HEV lesion in a stem vessel with intimal overgrowth and fragmented red cells. (H&E, 20x)
There was a high prevalence of some degree of villitis. Half of the placentas were from stillborn infants, but the existence of the same lesion in live born infants argued that it was not an artifact. Such microangiopathic changes had not been described in an early study of placental lesions in stillbirth. The placentas in Dr. Sander’s study were all referrals and would have been expected to have included those from stillbirths, but the paper did not report the percentage of all placentas in the registry that were from stillbirths. Less emphasized, but noted in that paper, was that vascular thrombosis was common in fetal vessels, and 14 of 16 cords were abnormal in the stillborn infants including “redundant” cords and 5 that were wrapped around fetal parts. Later papers correlated HEV with stillbirth, growth restriction in utero, villitis and other clinical outcomes that we will discuss later in the context of fetal vascular malperfusion. ,HEV would prove to be part of this malperfusion spectrum. Dr. Sander did note that areas of complete villous fibrosis (avascular villi) occurred distal to more proximal occlusive lesions. Dr. Sanders also illustrated in larger vessels “luminal septation or ‘bridging’ resulting from proliferation of intimal or subintimal cells” [17].
Dr. Sander also described a capillary variant of HEV that occurred in the small end branches of the villous tree. The pathologist can usually spot these areas microscopically at low magnification as slightly edematous and smudged villi (Fig 16a,b).
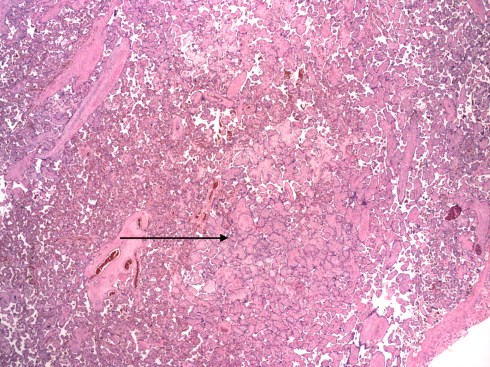
Fig 16a: This low power of the placental villi demonstrates an area with larger appearing, paler villi (arrow) compared to the surrounding normal villi due to capillary HEV. (H&E, 2x)
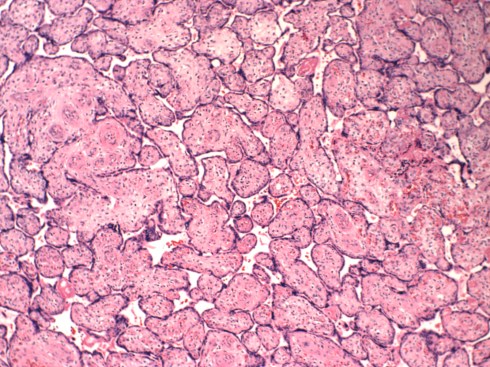
Fig 16b: This higher power of the large pale villi. (H&E, 10x)
Higher magnification demonstrates the hallmark lesion of red cells sheared into fragments by their passage through the capillary endothelium and deposited in the loose villous stroma (Fig 17).

Fig 17: This even higher power of villi from the same area as figure 16 shows the blue nuclear fragments of karyorrhexis and fragmented red blood cells. (H&E, 40x)
Current terminology emphasizes the endothelial and stromal karyorrhexsis (apoptosis of nuclei). The end result of occlusion of flow to these end villi is the loss of capillary walls, and an increase in collagen throughout the previously loose stroma creating dense pink empty villi under the microscope, designated avascular villi (Fig 18).

Fig 18: The villi in this picture have an eosinophilic (pink) stroma and absent capillaries. The small villi in the center show blue lines (ferrugination) just below the surface that is due to the deposition of iron transported by the syncytiotrophoblast to the stroma in the absence of circulatory transport back to the fetus. (H&E, 20x)
Perhaps the first use of the term avascular villi in English was by Dr. Peter Greunwald* in a 1961 paper in the New York State Journal of Medicine[21]. The paper was focused on a new topic that will be discussed in a later page, the infant who is small for gestation. Dr. Gruenwald noted the placental abnormalities, with two showing fetal vascular thrombi, and distal avascular villi. His description states“ If in a given area the blood pressure in the fetal circulation drops, perhaps by obliteration of the supplying artery, the pressure of the maternal circulation in the intervillous space may compress the fetal capillaries and cause their collapse.” This suggests that despite initial maintenance of luminal pressure, stromal edema collapses the capillaries.
* Footnote: Dr. Peter Greunwald was a pathology professor at Hahnemann Medical College who I recall giving a defiant lecture to our class on perinatal pathology. We had a new curriculum that compressed the basic medical sciences into one year, and Dr. Greunwald’s three hours had been reduced to one scheduled hour. Since it was the end of the day, Dr. Greunwald gave his three-hour presentation anyway. It is the only set of lecture notes from medical school that I still have.
A turning point in understanding of HEV was a simple experiment performed by Dr. Meredith Silvers and colleagues who cultured villi without blood flow in the fetal circulation, and noted lesions similar to HEV in the villi after 1 to 7 days[22]. The similarity was not identical but there was fragmentation and diapedesis of red cells. I found this at first confusing since there was no explanation for a pressure forcing the red cells through the vascular endothelial lining. However, the study also showed sclerosis of vascular lumens that may be simply the result of complete contraction of the muscular wall of the vessel. If this process was similar to that in the umbilical arteries with delivery, it is at least conceivable that cutting up pieces of placenta for tissue culture caused contraction of vascular media that in some segments of vessel trapped blood under pressure or rather than extravasating from the villi. In the umbilical artery, the generalized contraction of the artery after birth traps blood that causes beaded dilatations in the artery known as nodes of Hoboken. I suspect, based on the Cincinnati experience with fetal lambs on bypass having markedly increased placental vascular resistance that the deeper muscular vessels contract with loss of blood pressure. Likewise the post delivery transfusion with delayed cord clamping could in part be due to contraction of fetal arteries in the placental villi. This would only work if the contractions tended to start at the smaller vessels in the villi and progress to the larger vessels. This is all speculation, but vascular contraction might explain outward pressure on the red cells in Dr. Silver’s results. Dr. Silver’s study used two placentas with HEV distal to chorionic vein thrombi and a set of placentas from stillborn infants. In the stillborn infants, HEV developed in the vessels with blood and fibromuscular sclerosis in vessels without blood. (Fibromuscular sclerosis was demonstrated by electron microscopy to be an artifact of vasocontstriction and formalin fixation[23]. ) The cultured placentas did not develop the other features of HEV: myointimal proliferation, luminal septation, and hemosiderin deposition. They attributed this lack perhaps to the short duration of the experiments.
The study of avascular villi, the vascular changes of stillbirth and the in vitro culture of placental fragments, all pointed to the conclusion that HEV was a response to occluded fetal vascular flow in the placenta. A prospective study of placentas with HEV at the University of Pittsburgh, done after the above studies, found HEV in 13 of 1,938 placentas from unselected pregnancies, meaning these placentas had not been examined because of a complication of pregnancy[24]. In the HEV placenta group 6 mothers had prolonged deep variable decelerations, a marker of umbilical cord compression. Significant associations were present with nuchal cords in the infants, placental villitis and with fetal thrombi in placental vessels. The authors suggested “A reduction in umbilical blood flow may be a common link between hemorrhagic endovasculitis of the placenta and its associated maternal and fetal complications.”
The unified concept of fetal thrombotic vasculopathy was developed by Dr. Redline, a major innovator in the field of placental pathology, starting with a paper published with Dr. Pappin in 1995. The study defined specific criteria for a category of extensive avascular villi. These criteria were met in 29 placentas, approximately 1% of placentas examined, and correlated strongly with clinical complications in labor including variable heart rate decelerations and major thromboembolic events in the infants (4 with central nervous system infarctions and 1 with multiple organ infarctions). Five mothers had thrombophilia, but this did not rise to statistical significance against controls. At the time, I took the wrong messages from this paper. I only considered extensive avascular villi as important in writing my diagnostic reports. Fewer vascular villi were not given any diagnostic significance nor did I return to the placenta for further samples. I also assumed that the FVM was due to a thrombophilia transmitted to the fetus genetically or through the passage of gamma-immunoglobulin in anti-phospholipid disease. I thought that the placenta might well be a kind of thrombotic stress test with low flow and high turbulence, and that by examining all placentas, a higher incidence of thrombophilia might be uncovered. We will discuss the significance of thrombophilia in a later section. In retrospect I think both of these approaches were wrong. I overlooked the importance of the title of the paper “Fetal Thrombotic Vasculopathy” and that 17 of the placentas had thrombi, often non-occlusive.
One problem with many published medical papers is that the need to find an aggregate statistical significance results in some details being excluded from those papers. In Dr. Redline and Pappin’s paper referred to above, they did not report if there was a direct anatomic relationship of the thrombi with the avascular villi. They did not identify which lesions occurred in which thrombophilic mothers or infants with specific outcomes. These direct case to lesion connections cannot be found or verified statistically, yet they provide evidence to link chains of causation. The paper noted the association of thrombi and HEV with avascular villi, and that the avascular villi tended to occur in the areas of thrombi. The paper’s discussion, not to mention the title of the article, suggests that the authors saw a likely causal relationship of thombi, to HEV, and avascular villi, and this led to the paper below.
Dr. Redline and colleagues, including Drs. Sander, DeSa and Kraus, proposed a standardized terminology or nosology for the complex of lesions that they designated “Fetal Vascular Obstructive Lesions”[25]. This classification system, like other efforts to standardize terminology by the Society for Pediatric Pathology using validation of the concordance among pediatric pathologists, was intended to provide a standard nosology for further research into clinical correlations and causes of thrombotic disease in the placenta. They looked at criteria for both proximal thrombotic lesions, and for the distal villous lesions that corresponded to capillary HEV. Their final recommendation was to designate “changes consistent with chronic vascular obstruction, either severe (>15 involved villi/slide) or not otherwise specified.” The involved villi were to be averaged over the sampled slides, counting equally both avascular villi and those with stromal vascular karyorrhexis. The system makes some changes in diagnostic terms, but perhaps the most significant suggested change was to designate as Fetal Thrombotic Vasculopathy (any placenta with greater than 15 affected villi per slide using a random sample of three slides. Note: The term Fetal Thrombotic Vasculopathy was replaced by the term Fetal Vascular Malperfusion by the International Consensus group of placental pathologists[26].
As a practicing pathologist, I take a different approach to FVM. If fetal vascular obstructive lesions are first identified on the microscopic slides, our pathology group’s protocol was to reexamine the gross surface of the placenta for any suspicious vessels to sample (Fig 19a,b), and then relook at the cut slices for pale areas in the parenchyma to sample for avascular villi.
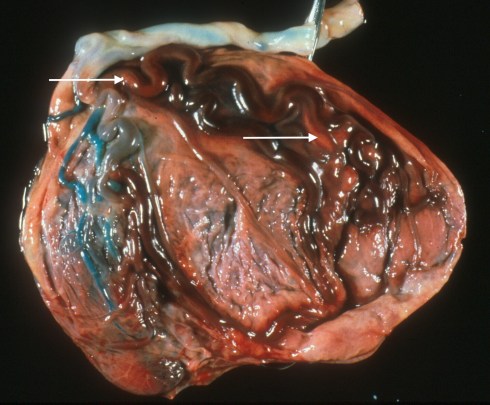
Fig 19a: An umbilical artery was injected with blue dyed barium and demonstrates the small area of placental still being perfused. The large red vessels are those involved in FVM. The arrows point to areas of mural thrombi.

Fig 19b: The surface of this placenta shows a single slightly red stained artery (arrow) compared to the adjacent arteries in an area of FVM.
The pallor can be more evident after the sliced placenta has been sitting and draining intervillous blood that may have obscured the bloodless avascular villi (Fig 20a,b).
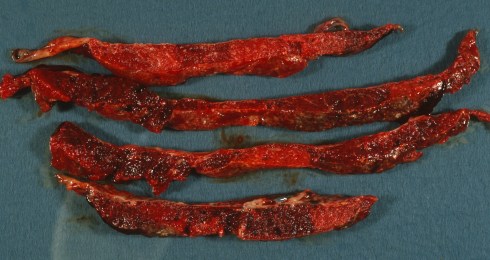
Fig 20a: These slices of a placenta demonstrate pale areas that proved to be due to HEV, FVM that likely would not have been as clearly seen with the fresh cut placenta.
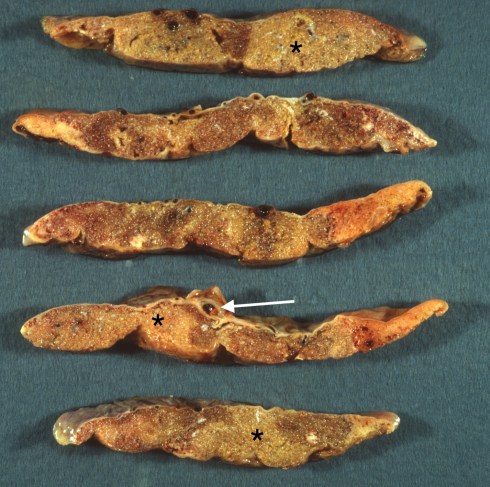
Fig 20b: These slices are fixed in formalin and show the pale areas very clearly (*) with one large stem vessel thrombus evident at the arrow.
A technique that I have not personally used that should aid identification of FVM is to stain the gross slices for 20 minutes with Perls Prussian blue solution [27]. This solution turns blue where there is ferric iron in the tissue. In FVM, it stains hemosiderin from broken-down blood cells in macrophages, and ferruginated villi. This latter iron accumulation occurs with absent, or even just decreased, fetal blood flow. Normally, the syncytiotrophoblast lining the villi actively transports transferrin-bound iron to the stromal side for transport by the fetal blood to the infant. The iron in the unperfused villi is like cargo being left on the pier for lack of transportation outward to the countryside. This resampling gives a more accurate description of the extent of villous thrombi and avascular villi. My personal, but not generally accepted terminology, was to diagnose Fetal Thrombotic Vasculopathy if there are thrombi as well as HEV or avascular villi. I used fetal occlusive vasculopathy (now fetal vascular malperfusion, FVM) for those placentas with only “downstream lesions”. Perhaps a more consistent approach is to call the lesions FVM but describe the presence and distribution of thrombi.
Current classifications of FVM make a distinction between partial global and segmental complete with the former describing diffuse change in distal villi often at the basal plate[28]. Segmental complete refers to the findings that would occur with an occlusion in a proximal chorionic or stem vessel. The extent of these lesions could vary since the more proximal an occluded chorionic vessel the more stem villi that will loose circulation. An occlusion in a single stem villous would affect all of the distal villi in that villous tree, and would appear as a coherent cluster of affected villi. Multiple discrete occlusions would demonstrate a pattern of affected villi that could not be accounted for by a single proximal occlusion. These distinctions may have etiologic and prognostic significance. I proposed a study in a blog in which FVM placentas by surface mapping, serially sectioning and reconstructing the vascular tree would specifically locate where thrombi formed and the volume of villi affected. The aim of the study was to analyze the “geographical” pattern of fetal thrombi in the placenta to discover anatomic and clinical features that would correlate/explain the specific location of thrombogenesis. Is there a pattern of thrombi that can be related to sharp angle branches or narrower chorionic vessels? Can any pattern be related to the timing or the severity of decreased umbilical blood flow? Would it be possible to distinguish thrombophilia from hypoperfusion? Perhaps it is too tedious and expensive a study for the expected benefits.
Stillbirth versus FVM
From the beginning HEV was correlated with stillbirth. This relationship could be an artifact. Even before HEV was described, it was reported that the fetal blood vessels in stillborn infants showed a proliferation of subintimal fibroblasts into the vascular lumen[29]. This is not surprising, since stillbirth causes a loss of fetal blood flow, but not of maternal blood flow that is sustaining the viability of the villi. These are almost the same conditions that occur in the villi distal to an occluded stem vessel. From the placental perspective, fetal death is effectively a total fetal occlusive vasculopathy. If we want to understand the relationship between FVM and stillbirth, we need to distinguish this intrinsic artifact due to fetal death from premortem. There are three differences that might aid in making this distinction. One is that FVM has back-pressure from the viable fetal circulation through the non-occluded vein or artery paired with the obstructed stem vessel. However, as we discussed in the experiment with minced villi, this back pressure may not lead to identifiable changes in the distal lesions. A second very diagnostic useful difference is that thrombi, which require blood flow to form, cannot occur after fetal death. Not all placentas with evidence of FVM have identifiable thrombi. The third difference is that in fetal death the process should involve all of the villi simultaneously, while FVM occurs in one vessel at a time. A combination of early changes such as karyorhectic debris in some villi and avascular villi in others suggests FVM before death. This was an observation that Dr. Fox made in his early paper on chorionic artery thrombi. In practice, the diagnosis of such heterogeneous involvement can be complicated since as we will see, there is some intrinsic variation in timing of endothelial ingrowth even after fetal death, and the end stage of villits is also avascular villi. The ability to distinguish premortem FVM from postmortem change is necessary to show a true correlation of FVM and stillbirth. We will consider in a subsequent section the evidence not only that FVM is correlated wtih stillbirth, but how to interpret the complex causality of that relationship.
The idea that there is a progression of the distal lesions in FVM is reasonable, but cannot be proven by observing placentas that are delivered and examined in a single time frame. Unlike for example studies in myocardial infarction, we do not know when the occlusion occurred in a particular stem villous. In stillborn infants we can estimate the timing of the cessation of fetal blood flow since this is the interval of intrauterine postmortem retention. We would expect that the changes of FVM should occur in all villi and at a uniform pace. Thus, the changes in the FVM lesions would have the same timing in a given infant as the postmortem retention.
There are limits to the pathologist’s ability to time the intrauterine postmortem retention interval because we seldom know the exact time of the intrauterine death. However, pathologists have developed techniques to estimate the intrauterine postmortem retention interval because of its importance to understanding of the timing and causes of stillbirth. This interval was often estimated in the past by the degree of maceration (softening) of the infant. There was an objective progression of changes, but the timing of these changes was an estimate. (An aside, maceration is also a cooking term for soaking an ingredient in a liquid to soften it, a grisly analogy. I prefer not to use the term.) When I was a resident in 1975-76, we often relied on an x-ray finding of overlapping skull bones (Spaulding sign) as evidence of more than 48 hours of postmortem retention. This was an act of faith.
Then in the 1980’s, Dr. David Genest and colleagues published a landmark series of papers that times retention of the fetus in the uterus after death and before delivery[30-32]. The obstetricians had provided the last time the infant was known to be alive and first time the infant was proven to be dead based on detecting fetal heart beat. This data provided a window in time in which the fetus had died prior to delivery. The pathologists reviewed the photographs of the stillborn body, the microscope slides of the placenta and the microscope slides of the body organs. The latter provided the most accurate estimate of the timing of the infants’ deaths. This approach was based on evaluating the autolysis that occurs when cells die from a lack of oxygen. In this process, initially the lack of oxygen results in anaerobic metabolism with accumulation of lactic acid. The beginning of this phenomenon is familiar to those whose muscles cramp from extreme exercise. After exercise reoxygenation occurs, but with death the blood supply ceases, the cells become progressively more acidotic until the low pH becomes a signal for the release of digestive enzymes (lysozymes) from lysosomes (membrane bound vesicles) whose purpose is to initiate autolysis. In a living organism autolysis may form a useful purpose such as removing dead tissue to allow regeneration or scar formation. In the stillborn fetus the process of autolysis occurs under uniform and ideal conditions in that the fetus is in a body temperature water bath, the optimal temperature for lysosomal enzymes to act and is free of normal bacterial flora that can digest cells. (Bacterial infection can cause or be associated with stillbirth, but it usually initiates labor creating a short postmortem retention). A pathologist recognizes autolysis under the microscope as coagulation necrosis using the routine stain of hematoxlyn (blue) and eosin (pink). The lysosomal enzymes remove the blue color (basophilia) from the nucleus and smudge the organelles in the cytoplasm to a bright homogenous pink. Fortunately for timing estimates, the cells of different organs proceed at different paces in producing visible coagulation necrosis. Using some test cases, Dr. Genest and colleagues were able to show that 1) the difference between no loss of nuclear basophilila, loss of some basophilia and the loss of all basophilia could be reliably distinguished and 2) the loss of basophilia in a subset of tissues correlated well with the known windows in which death occurred. For example if there was some loss of basophilia in the tubules of the kidney, it was present in all fetuses whose windows indicated more than 4 hours of death before delivery, including, 4-24 hours, 24-48 hours, 48-96 hours, 1 week, 3 weeks, but not in fetuses known to have died before 4 hours of birth. Dr. Genest recognized that errors might occur in certain situations for example shock prior to death can lead to coagulation necrosis in the kidney and liver, or that keeping the body in a warm environment, for enough time, would permit some autolysis to continue after birth. While not a perfect method, this is still the best approach that we as pathologists have for determining the time of death in utero.
Genest and colleagues using the same patients and techniques as in the autopsy study evaluated progressive changes in the placenta and in body photographs with the fetal retention intervals. The placental features were less accurate than the autopsy histology. These studies were undertaken and were useful because not all stillborn infants are autopsied.
The earliest finding in the placenta that occurs only hours after fetal death is karyorrhexis of nucleated fetal blood cells (Fig 21).
This nuclear fragmentation is evidence of apoptosis resulting from a stimulus that leads to a cellular cascade that has been likened to cell suicide. Temporally, the next change that develops is the progressive septation by fibroblast ingrowth into the lumen of villous stem vessels (Fig 22).

Fig 22: This stem vessel from a prolonged postmortem intrauterine retention in a twin shows fibroblast ingrowth into the clotted blood. The red cells are not fragmented as in HEV, but this has not been demonstrated to be a reliable feature to distinguish premortem from postmortem fetal vascular obstructive changes. (H&E, 20x)
The last stage is the development of avascular villi. The precision of this scheme for timing has been questioned studying the placentas of feticide in the treatment of complications multiple gestation pregnancy, but the validity of the progression is obvious to pathologists that perform stillborn autopsies[33]. After the publication of Dr. Genest’s study, Dr. Sander published a study that subdivided the categories of HEV as active, bland or healed and as diffuse or focal[20]. The bland form correlated with the duration of stillbirth retention in the uterus. A prospective blinded study has not been done evaluate HEV like changes as predictors of stillborn versus livebirth. Dr. Genest study does provide evidence that it takes (days) to develop HEV like changes, and weeks to develop avascular villi.
Causes of fetal vascular thrombi
Genetic thrombophilia
The causes of thrombi formation in any vessel were established by the founder of cellular pathology and are hence known eponymously as Virchow’s triad. The triad is hypercoagulability (thrombophilia), blood flow stasis, and vessel injury.
Considered from an evolutionary point of view, the ease of coagulation of the blood is a double-edged sword. On one hand clotting quickly can prevent bleeding to death if injured or attacked by a predator. On the other hand easy clotting can causes thrombi in vessels producing stroke or myocardial infarction. The proteins, and hence genes, controlling the coagulation of blood are very complex in order to keep a balance of clotting versus blood flowing. Not surprisingly, there are multiple genes in the population that are thrombophilic such as mutations in the genes for blood clotting factors such as Factor V (the Leiden mutation), prothrombin, methyl tetrahyrdate folate reductase, antithrombin III, and others. Generally thrombophilic genes are heterozygous in the individual, that is one copy of the mutated gene, and are not or only weakly associated with fetal thrombotic vasculopathy[34, 35]. For example, in a prospective study of mothers who were heterozygotic for the Factor V, Leiden mutation, a common thrombophilic mutation, there was a statistically significant increase of avascular villi in the 50 infants who were heterozygous for the mutation, but not for fetal thrombotic vasculopathy[36]. Another study demonstrated fetal stem vessel thrombi in 7of 13 women with confirmed thrombophilia and pregnancy complications (4 of these were stillbirths) [37]. If a thrombophilic gene in the infant is homozygous, then thrombi in the placenta and infant have been reported. Interestingly, the combined effect of two different mutations on the same thrombophilic pathway can cause placental and fetal thrombotic disease. Such was a case that my colleagues and I presented in grand rounds.
Autoimmune thrombophilia
Some mothers, usually with a generalized autoantibody dysfunction such as systemic lupus erythematosis, have false positive tests for syphilis and prolonged clotting in the test tube measure of prothrombin time. This was initially named lupus anti-coagulant despite its association with thrombophilia in the body and with an increased incidence of spontaneous fetal abortion. The antibody causing the abnormal testing was first identified as anti-cardiolipin, improbably an antibody to an antigen on the inner mitochondrial membrane. This observation was expanded to show that the anti-cardiolopin antibodies recognized phospholipids in the cell wall and could induce blood clotting in living blood vessels. When my colleagues and I looked prospectively at mothers with measurable anti-cardiolipin antibodies, the placentas were normal[38]. In other reports anti-phospholipid antibodies have been associated with multiple placental infarctions. (Infarctions are discussed in another chapter, but they are attributable to thrombus in maternal spiral arteries not the fetal vessels. Our group reported a case of a mother admitted with a stillborn infant, who died overnight in the hospital from widespread thrombi in the heart, kidney and brain[39]. The placenta had infarctions all of the same duration that were coeval with the postmortem retention interval of the fetus.
The placenta transports maternal immunoglobulin G to the fetus. In some infants fetal thrombotic vasculopathy can be attributed to the transport of thrombophilic antibodies[40]. Anti-phospholipid antibodies have been found as the likely explanation of rare cases of fetal thrombotic vasculopathy including in a stillborn infant[41].
Inflammation
Multiple observers have found an association of villitis with avascular villi and even with chorionic vascular thrombi[10, 42]. The villitis typically is lympho-histiocytic in composition and does not have a known infectious etiology. The association of villitis and its association with fetal growth restriction will be discussed in more detail in a later chapter. In this section, the relevant aspect is that Dr. Paige Faulk and colleagues demonstrated a mechanism relating villitis to fetal thrombotic vasculopathy[43]. They developed antibodies to key antigens of coagulation as part of their studies comparing immune rejection between placentas and organ transplants. They applied these antibodies to frozen sections of placentas with villitis and demonstrated an up-regulation of coagulation factors in fetal capillaries similar to the changes in transplant rejection. Under the microscope the pathologist usually sees only the consequences of vaso-destruction, but small thrombi can sometimes be found in the inflamed villi (Fig 23).
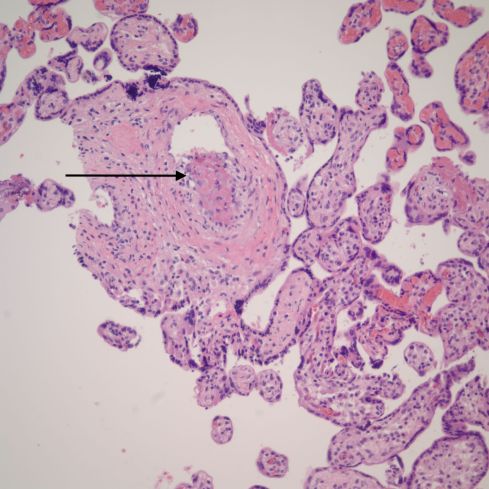
Fig 23: The villi in the center demonstrate lympho-histiocytic villitis with loss of fetal vessels and a mild increase in inflammatory cells. The arrow points to a thrombus in a small vessel. (H&E, 20x)
These thrombi are likely a direct consequence of thrombophilia induced by the lymphocytic inflammation.
Acute inflammation of the fetal vascular system can also cause thrombosis likely from direct inflammatory injury of the endothelium. This type of mural fibrin deposition in direct contact with inflammation in surface chorionic vessels has been well described[44]. This lesion however is not identical those described in FVM and given the association of such acute infection with preterm labor and delivery, this is not a precursor of FVM. A possible correlation with inflammation of umbilical vessels, particularly necrotizing inflammation, is less well established. An interesting observation that I have seen multiple times is a necrotizing funisitis (more in the chapter on infection) in which the necrosis is between a completely thrombosed artery and the neighboring vessels. This lesion is almost certainly a reaction to the thrombosed artery and surrounding necrosis of the wall rather than a cause of the thrombosis (Fig 6a)
Maternal diabetes
Another possible cause of thrombophilia occurs in mothers of diabetic infants. Dr. Fox’s early paper on fetal vascular thrombosis noted a high incidence in infants of diabetic mothers[10]. Such infants have a well-documented increase in renal vein thrombosis, perhaps related to the hyperosmolar urine from fetal hyperglycemia[45]. I have not seen FVM with infants of diabetic mothers, perhaps because of better treatment of diabetes in pregnancy.
Vascular stasis and trauma
The majority of placentas with FVM due not have an associated thrombophilia in the infant. From Virchow’s triad, that leaves stasis and vascular injury as possible causes. Dr. Redline using his criteria for fetal thrombotic vasculopathy examined 125 placentas from medical legal consultations with infants with severe neurologic lesions[46]. He found 23 placentas meeting the criteria, and of these there was a statistically significant, high incidence of umbilical cord entanglement and of other umbilical cord abnormalities including thin cords, highly coiled cords, and marginally inserted cords compared to the cases without FVM criteria. This made sense if the umbilical cord abnormality, for example wrapping, caused local injury to the endothelial lining of the vessel from compression or torsion, and slowed blood flow. These changes, per Virchow’s triad, would favor thrombi in the fetal circulation much as an injured limb in a cast favors thrombi. The slowed flow in the umbilical vein would be most susceptible to thrombi, like the deep leg veins with stasis from prolonged sitting. The association of umbilical cord and fetal vascular thrombi has been confirmed in other studies and case reports[47, 48]. Our understanding is incomplete. The mechanism of stasis in umbilical vein flow does not directly explain why the distribution of thrombi in chorionic surface and villous stem vessels.
Other possible mechanisms of thrombosis include direct fetal trauma or fetal vascular constriction. The first explanation was suggested by Dr. Naeye, who imagined fetal kicking of the placenta as a possible cause. The pressure of fetal part against the placenta might also injure and compress a surface vessel leading to thrombosis. I have seen patterns of thrombi that anatomically suggest a single area of pressure on the placenta, but I have no way to prove that etiology after delivery (Fig 24).
Fig 24: A smooth margin of pallor from occluded stem vessels in the placenta from marginal compression? (H&E, 20x)
We have already discussed the unique ability of the umbilical arteries to constrict permanently after delivery, and of the placental circulation to become irreversibly high resistance after fetal by-pass for experimental surgery in sheep. A role for arterial constriction in producing thrombus cannot be excluded. Turbulence because of sharp angles in the fetal circulation may also play a role. While some cushions may form in a normal circulation, certainly they provide an area more sensitive to thrombogenic mechanisms. Figure 13a in the lower right corner shows that the thrombus is occurring above and at a point where a branch is diving below the surface. One unexplained possible association was the report of FVM in three surviving dizygotic twins who were co-twins with a complete hydatidiform mole[49]. I have only seen one such twin case personally, and I failed to observe FVM.
The clinical significance of FVM
Intrauterine growth restriction
Starting with Sander’s first paper, HEV was prevalent in intrauterine growth restricted, or small for gestation, infants. This topic of growth restriction will be discussed in more detail in a later chapter. For this chapter, it is sufficient to understand that if the fetal blood supply is occluded permanently to portions of the placenta, then functional placental tissue will be lost. At some point, this loss of function cannot be compensated, or stated another way the placental reserve is exceeded. Experience suggests there are three different limits to that reserve. The most severe loss compromises fetal respiration and can result in hypoxia and acidosis. We will consider this in discussing asphyxia as a cause of stillbirth in growth restricted infants. The second limit is the point at which the placenta can maintain respiration, but this comes at the cost of fetal nutrient transfer. We will discuss this in the chapter on chronic intrauterine growth restriction. Finally, there is the placenta that can meet the daily needs of the infant, but does not have the reserve to recover quickly from uterine contractions or any other insult that might require increased respiratory function. This is analogous to an adult with chronic lung disease who is not hypoxic at rest, but cannot sustain intense exercise or increased metabolic demand. In the fetus, this level of compromise of the placenta could lead to hypoxia with labor. As pathologists examining the placenta, we are not able to assign the compromised placenta to one of these categories based purely on the anatomic findings. This failure in part is due to the inability to quantify functional reserve that will be discussed in the chapter on growth restriction. Better pathological techniques could be developed to measure placental functional reserve. These measures would then need to be evaluated in relation to the fetal metabolic needs, the events of labor, the fetal blood flow, and other variables that would affect the clinical outcome. Within these limitations, an estimate of the volume of avascular villi is a rough measure of lost placental function that can be included in the pathological evaluation of HEV. As we will see, FVM may be an important marker of decreased umbilical cord blood flow. This decreased blood flow likely explains the correlation of FVM with fetal distress in labor and with stillbirth better than loss of functional placental tissue. The relative role in compromising fetal growth of intermittent or partially decreased umbilical blood flow compared to loss of functional placental tissue has not been resolved.
Fetal stroke
When I began my career in obstetrical pathology, there was little concern about thrombi in fetal placental vessels. An arterial thrombus would simply devitalize a small area of placenta, causing a loss of function in that small volume of placenta. The placenta appeared capable of absorbing such injury without permanent damage to the fetus. The possibility of thrombo-embolic fetal brain infarction (as opposed to in situ thrombi in severe thrombophilia) was not considered until better neonatal brain imaging (MRI) demonstrated such infarctions. As a resident, I autopsied a newborn infant who demonstrated multiple discrete hemorrhagic infarctions in the cerebral cortex, and my mentors had no explanation for this pattern. Later as a fellow in pediatric pathology, the autopsy of a newborn infant demonstrated a massive cerebral infarction in the territory of the middle cerebral artery. This is the same lesion seen in a typical adult stroke that causes one-sided paralysis. The consensus of my mentors at the time was that the stroke was due to fetal asphyxia and that there must have been a narrow middle cerebral artery as an incidental anomaly. This seemed like an improbable explanation even at the time.
In 1993, Drs. Kraus, and Rayne published a paper arguing that the finding of a thrombus in the fetal circulation was evidence that thrombosis in the fetus was possible, and that such thrombi might be associated with brain lesions[50]. The paper noted that most placental thrombi are in the surface veins, but did not elaborate on the causal connection between FVM and neonatal cerebral infarction. My understanding of these stroke cases was crystalized by a follow up paper of Dr. Fred Kraus based on 15 medico-legal placental pathology consultations for infants with cerebral palsy[51]. He found thrombi in 11. One case had an autopsy and thrombi and infarctions were found in the brain. He speculated that other cases of cerebral palsy might be from either embolism or in situ formation of thrombi in which there were thrombi in the placental vessels.
In retrospect, there was good reason to expect fetal strokes with FVM based on the fetal circulation. In the fetus, oxygenated blood arrives at the heart from the umbilical vein, passes through the foramen ovale into the left ventricle and then out the aorta. Most of this oxygenated blood goes directly to the arteries supplying the heart and brain that branch before the ductus arteriosus. Most of the un-oxygenated blood from the body leaving the right ventricle travels from the pulmonary artery across the ductus arteriosus (a small amount traverses the high resistance vessels of the lung) mixing with oxygenated blood in the descending aorta and going to the rest of the body and placenta. The good result is that the heart and brain are well oxygenated, but this arrangement favors any thromboembolism (a piece of thrombus broken free from a mural thrombus) finding its way to the brain. These emboli are likely to be of a size to occlude a cerebral artery causing loss of blood flow and infarction (death) of the brain tissue relying on that artery. Unlike the symptomatic strokes of adults, cerebral infarctions in infants may be clinically silent, and the infants often recover without notable deficit.
Now, if FVM is identified, before or after the placenta has been examined pathologically, an attempt should be made to determine if there is a thrombus in a surface chorionic vein. (As noted, arteries cross over the veins, and vessels with stasis may have a red discoloration from diffusion of hemoglobin from ischemic red blood cells.) Rarely brain infarctions in neonates will be due to a severe thrombophilia causing thrombi to form in situ in the brain, or to embolize from a vegetation on the heart, but the majority are due to FVM on the umbilical venous side.
This example case of neonatal stroke occurred in a term, normal weight infant who had some fetal distress with Apgar scores of 4 at 1 minute, 6 at 5 minutes, and 7 at 10 minutes of age. The placenta had numerous fetal vascular thrombi (Fig 25)

Fig 25: The asterisks demonstrate thrombi in a stem vessel in the placenta from the described case. (H&E, 20x)
. A head CT demonstrated hemorrhages in the left frontal and right posterior parietal areas. His father was simultaneously admitted 2 weeks later for deep vein thrombosis and was found to have the Leiden V mutation. The infant’s mother had an anti-thrombin III deficiency. On testing, this infant was heterozygous for both of these causes of thrombophilia which were additive. Since he did not develop other thrombotic problems in the nursery, the brain lesions may well have been embolic from the placenta.
This case demonstrates how an uncomplicated pregnancy with no, or minor, fetal distress at delivery might result in subsequent cerebral palsy. Some of these infants have brain lesions suggestive of intrauterine stroke on early radiologic studies. In a report of six such infants in which the placentas were studied, one placenta demonstrated a resolved retroplacental hematoma and the other a dysmature small placenta[52]. The mother of the infant with the dysmature placenta had a history of thrombophlebitis treated with heparin. Given the difficulty of recognition and sampling with FVM, some of unexplained cerebral palsy with porencephaly (discrete volumes of lost brain tissue) might have had placental thrombi.
Possibly a very large prospective study of placentas with FVM would demonstrate an association with the rare case of silent neonatal stroke given the logical mechanism. Informally, our study of several thousand unselected placentas did not demonstrate any cases. Whether a neonate would benefit from further diagnostic evaluation of the brain if the placenta shows FVM, particularly in large chorionic veins, requires further study.
FVM and liver disease
Another possible consequence of FVM was reported on a total of 4 infants with severe neonatal liver diseases and FVM[53, 54]. Three of the infants had cerebral infarctions. One had a hepatic vein thrombosis. This thrombus in continuity with the umbilical vein is plausibly part of the same process. The three cerebral infarctions are consistent with a high incidence of fetal thromboemboli in FVM implying that some of the thrombi were on the venous side of the placental circulation. The etiologic relationship, if any, of FVM to the liver disease in the three cases without hepato-vascular thrombi was not found.
FVM associated with stillbirth
My experience and that of others suggests that the majority of Dr. Kraus’s medico-legal cases could not have had cerebral palsy due to cerebral infarction. His paper did note that 2 of the patients had long, double nuchal cords[51]. Dr. Redline like Dr. Kraus, but using his own database of medico-legal referrals found a highly significant prevalence of fetal thrombotic vasculopathy in infants with cerebral palsy, (more or less equally divided between those with and without criteria of neonatal encephalopathy) 23 of 125 (18%) compared to 6 of 250 (2%) controls [55]. These medico-legal cases were the same cases that provided the evidence for an association of umbilical cord abnormalities with FVM[46].
A study of Parast and colleagues concluded that in stillbirths, fetal vascular thrombi were a marker of umbilical cord accident with high specificity if they met criteria of vascular ectasia, thrombi in the chorionic plate or stem vessels and evidence of villous changes of fetal vascular obstructive lesions in the villi[56]. The reasoning was somewhat circular in that the pathologists initially had to assign the cause of death as due to a cord accident or some other mechanism, or to “unknown”. Those 10 cases prospectively assigned as cord accidents did show the criteria, as did 13 of 25 unknown cases and 3 of 27 cases of known, non-cord accident cases. Of the latter they suggest that one case attributed to heart disease in retrospect also could have been due to a cord lesion. They then discuss the problems with interpreting villous lesions with prolonged stillbirth, very acute cord compression without lesions, and the added confidence of also finding a cord lesion. For me at the time, the criterion for vascular ectasia, “vascular distention to at least four times the diameter of an adjacent muscular vessel of similar caliber”, was novel. No test of concordance among pathologists nor of the prevalence of such lesions in normal placenta were provided. This paper was provocative for showing such a high prevalence in stillbirths of suspected umbilical cord accident and of unknown cause.
Some of the same authors and others published a study the next year that compared the same fetal vascular lesions in the placentas of living infants with and without cord lesions[57]. The gross cord lesions included long cords, cord knots, wrapped cords, marginal or velamentously inserted cords, twisted and thin cords. In agreement with earlier research these complicated cords had significantly more fetal vascular lesions and more neonatal complications. However, even with strong statistical significance, the prevalence of vascular ectasia was high in controls and cases 59% versus 78%. The authors duly noted the shortcomings of their excellent study, which was hampered by being retrospective. They attempted another retrospective study that compared 113 placentas that had been diagnosed with features of fetal thrombotic vasculopathy to 216 contemporary controls[58]. Interestingly this study did not find an association with gross umbilical cord abnormalities, but did confirm the association of FVM with stillbirth and non-reassuring fetal heart rate recordings. The inclusion of preterm placentas and of only retrospective clinical information might account for the lack of association with umbilical cord abnormalities. New associations were found with oligohydramnios and fetal cardiac anomalies. The former would hark back to the known association of oligohydramnios and umbilical cord compression. The association with cardiac anomaly is interesting, but the causational relationship is speculative. Finally, they reconfirmed at another institution that stillborn infant placentas with cord abnormalities, defined as those known to be associated with FVM, had a very significantly higher prevalence of FVM compared to control stillborn infant placentas[59].
Is FVM a marker of Fetal Asphyxia in Cerebral Palsy?
The evidence from the stillbirth cases suggests that FVM is a marker for umbilical cord compromise that can eventually lead to fetal death. A logical leap is that the lesion might also be a marker for hypoxic ischemic neonatal brain injury.
This conclusion was suggests both in Dr. Sander’s correlation of brain injury with HEV and with Dr. Redline and colleagues’ work of the correlation with the extent of avascular villi as a predictor[17, 60]. Dr. Redline cites in a recent review an even more definitive study of this question that is to be published[28].
Conclusion
The evidence from the stillborn studies, the early association of HEV with evidence of fetal distress in labor, and the association of FVM with umbilical cord abnormalities, all point to a single hypothesis. FVM is often caused by umbilical cord compromise. As a marker for this compromise, it is evidence that obstructed umbilical cord blood flow accounts for the associations of FVM with stillbirth and fetal distress. Missing knowledge includes how the various cord findings affect the flow of umbilical cord blood in the living fetus and how this leads to the distribution of thrombotic lesions in the placenta.
Following the logic of Virchow’s triad, some cases of FVM will result from fetal thrombophilia, but this has proven to be a lesser factor than umbilical cord compromise. It remains to be shown how umbilical cord lesions produce the thrombosis. Is the vessel occlusion purely from intermittent stasis of flow, or ischemic injury of endothelium, or are the interactions more complex?
References
- Devlieger, H., et al., Thrombosis of the right umbilical artery, presumably related to the shortness of the umbilical cord: an unusual cause of fetal distress.Eur J Obstet Gynecol Reprod Biol, 1983. 16(2): p. 123-7.
- Solano Sanchez, S.R., et al., [Umbilical artery thrombosis. A report of a case and review of the literature].Ginecol Obstet Mex, 2005. 73(6): p. 332-5.
- Afriat, R., et al., [Prenatal diagnosis of spontaneous thrombosis of the umbilical artery during the third trimester of pregnancy. Two cases with surviving infants].J Gynecol Obstet Biol Reprod (Paris), 1995. 24(4): p. 411-4.
- Raio, L., et al., In-utero characterization of the blood flow in the Hyrtl anastomosis.Placenta, 2001. 22(6): p. 597-601.
- Hobel, C.J., et al., Ligation of one umbilical artery in the fetal lamb; experimental production of fetal malnutrition.Obstet Gynecol, 1970. 36(4): p. 582-8.
- Heifetz, S.A., Thrombosis of the umbilical cord: analysis of 52 cases and literature review.Pediatr Pathol, 1988. 8(1): p. 37-54.
- Klaritsch, P., et al., Spontaneous intrauterine umbilical artery thrombosis leading to severe fetal growth restriction.Placenta, 2008. 29(4): p. 374-7.
- Sato, Y. and K. Benirschke, Umbilical arterial thrombosis with vascular wall necrosis: clinicopathologic findings of 11 cases.Placenta, 2006. 27(6-7): p. 715-8.
- Cook, V., et al., Umbilical artery occlusion and fetoplacental thromboembolism.Obstet Gynecol, 1995. 85(5 Pt 2): p. 870-2.
- Fox, H., Thrombosis of foetal arteries in the human placenta.J Obstet Gynaecol Br Commonw, 1966. 73(6): p. 961-5.
- DeSa, D.J., Intimal cushions in foetal placental veins.J Pathol, 1973. 110: p. 347-352.
- Scott, J.M., Fibrinous vasculosis in the human placenta.1983: p. 87-99.
- Sander, C.H., Hemorrhagic endovasculitis and hemorrhagic villitis of the placenta.Arch Pathol Lab Med, 1980. 104: p. 371-373.
- Sander, C.H. and N.G. Stevens, Hemorrhagic endovascullitis of the placenta: an indepth morphologic appraisal with initial clinical and epidemiologic observations.Path Annual, 1984. 19: p. 37-79.
- Stevens, N.G. and C.H. Sander, Placental hemorrhagic endovasculitis: risk factors and impact on pregnancy outcome.Int J Gynaecol Obstet, 1984. 22(5): p. 393-7.
- Rayburn, W., et al., The stillborn fetus: Placental histologic examination in determining a cause.Obstet Gynecol, 1985. 65: p. 637-641.
- Sander, C.H., et al., Haemorrhagic endovasculitis of the placenta: a review with clinical correlation.Placenta, 1986. 7: p. 551-574.
- W, R., S. C, and C. A, Histologic examination of the placenta in the growth-retarded fetus.Am J Perinatol, 1989. 6:58-61.
- Novak, P.M., et al., Report of fourteen cases of nonimmune hydrops fetalis in association with hemorrhagic endovasculitis of the placenta.Am J Obstet Gynecol, 1991. 165(4 Pt 1): p. 945-50.
- Sander, C.M., et al., Livebirths with placental hemorrhagic endovasculitis: interlesional relationships and perinatal outcomes.Arch Pathol Lab Med, 2002. 126(2): p. 157-64.
- Gruenwald, P., Abnormalities of placental vascularity in relation to intrauterine deprivation and retardation of fetal growth. Significance of avascular chorionic villi.N Y State J Med, 1961. 61: p. 1508-13.
- Silver, M.M., H. Yeger, and L.D. Lines, Hemorrhagic endovasculitis-like lesion induced in placental organ culture.Hum Pathol, 1988. 19: p. 251-256.
- van der Veen, F., S. Walker, and H. Fox, Endarteritis obliterans of the fetal stem arteries of the human placenta: an electron microscopic study.Placenta, 1982. 3(2): p. 181-90.
- Shen-Schwartz, S., T.A. Macpherson, and E. Mueller-Heubach, The clinical significance of hemorrhagic endovasculitis ofthe placenta.Am J Obstet Gynecol, 1988. 159: p. 48-51.
- Redline, R.W., et al., Fetal vascular obstructive lesions: nosology and reproducibility of placental reaction patterns.Pediatr Dev Pathol, 2004. 7(5): p. 443-52.
- Khong, T.Y., et al., Sampling and Definitions of Placental Lesions: Amsterdam Placental Workshop Group Consensus Statement.Arch Pathol Lab Med, 2016. 140(7): p. 698-713.
- McDermott, M. and J.E. Gillan, Trophoblast basement membrane haemosiderosis in the placental lesion of fetal artery thrombosis: A marker for disturbance of materno-fetal transfer.Placenta, 1995. 16: p. 171-8.
- Redline, R.W. and S. Ravishankar, Fetal vascular malperfusion, an update.APMIS, 2018. 126(7): p. 561-569.
- Fujikura, T. and R.C. Benson, Placentitis and Fibrous Occlusion of Fetal Vessels in the Placentas of Stillborn Infants.Am J Obstet Gynecol, 1964. 89: p. 225-9.
- Genest, D.R., Estimating the time of death in stillborn fetuses: II Histologic evaluation of the placenta: a study of 71 stillborns.Obstet Gynecol, 1992. 80: p. 585-592.
- Genest, D.R. and D.B. Singer, Estimating the time of death in stillborn fetuses: III. External fetal examination; a study of 86 stillborns.Obstet Gynecol, 1992. 80(4): p. 593-600.
- Genest, D.R., M.A. Williams, and M.F. Greene, Estimating the time of death in stillborn fetuses: I. Histologic evaluation of fetal organs; an autopsy study of 150 stillborns.Obstet Gynecol, 1992. 80: p. 575-84.
- Jacques, S.M., et al., Estimation of time of fetal death in the second trimester by placental histopathological examination.Pediatr Dev Pathol, 2003. 6(3): p. 226-32.
- Mousa, H.A. and Z. Alfirevic, Do placental lesions reflect thrombophilia state in women with adverse pregnancy outcome?Hum Reprod, 2000. 15(8): p. 1830-3.
- Leistra-Leistra, M.J., et al., Fetal thrombotic vasculopathy in the placenta: a thrombophilic connection between pregnancy complications and neonatal thrombosis?Placenta, 2004. 25 Suppl A: p. S102-5.
- Rogers, B.B., et al., Avascular villi, increased syncytial knots, and hypervascular villi are associated with pregnancies complicated by factor V Leiden mutation.Pediatr Dev Pathol, 2010. 13(5): p. 341-7.
- Arias, F., et al., Thrombophilia: a mechanism of disease in women with adverse pregnancy outcome and thrombotic lesions in the placenta.J Matern Fetal Med, 1998. 7(6): p. 277-86.
- Bendon, R.W., et al., Prenatal screening for anticardiolipin antibody.Am J Perinatol, 1990. 7(3): p. 245-50.
- Bendon, R.W., et al., A maternal death due to thrombotic disease associated with anticardiolipin antibody.Arch Pathol Lab Med, 1987. 111(4): p. 370-2.
- Levine, J.S., D.W. Branch, and J. Rauch, The antiphospholipid syndrome.N Engl J Med, 2002. 346(10): p. 752-63.
- Brewster, J.A., S.M. Quenby, and Z. Alfirevic, Intra-uterine death due to umbilical cord thrombosis secondary to antiphospholipid syndrome.Lupus, 1999. 8(7): p. 558-9.
- Kraus, F., Case 3 Placenta: thrombosis of fetal stem vessels with fetal thrombotic vasculopathy and chronic villitis.Pediatr Pathol, 1996. 16: p. 143-148.
- Labarrere, C.A., J.A. McIntyre, and W.P. Faulk, Immunohistologic evidence that villitis in human normal term placentas is an immunologic lesion.Am J Obstet Gynecol, 1990. 162(2): p. 515-22.
- Redline, R.W., et al., Placental lesions associated with neurologic impairment and cerebral palsy in very low-birth-weight infants.Arch Pathol Lab Med, 1998. 122(12): p. 1091-8.
- Oppenheimer, E.H. and J.R. Esterly, Thrombosis in the newborn: comparison between infants of diabetic and nondiabetic mothers.J Pediatr, 1965. 67(4): p. 549-56.
- Redline, R.W., Clinical and pathological umbilical cord abnormalities in fetal thrombotic vasculopathy.Hum Pathol, 2004. 35(12): p. 1494-8.
- Taweevisit, M. and P.S. Thorner, Massive fetal thrombotic vasculopathy associated with excessively long umbilical cord and fetal demise: case report and literature review.Pediatr Dev Pathol, 2010. 13(2): p. 112-5.
- Chan, J.S. and R.N. Baergen, Gross umbilical cord complications are associated with placental lesions of circulatory stasis and fetal hypoxia.Pediatr Dev Pathol, 2012. 15(6): p. 487-94.
- Ernst, L.M., D. Chou, and S. Parry, Fetal thrombotic vasculopathy in twin placentas with complete hydatidiform mole.Pediatr Dev Pathol, 2009. 12(1): p. 63-7.
- Rayne, S. and F. Kraus, Placental thrombi and other vascular lesions classification; morphology and clinical correlations.Path Res Pract, 1993. 189: p. 2-17.
- Kraus, F.T., Cerebral palsy and thrombi in placental vessels of the fetus.Hum Pathol, 1997. 28: p. 246-248.
- Scher, M.S., et al., Destructive brain lesions of presumed fetal onset: antepartum causes of cerebral palsy.Pediatrics, 1991. 88(5): p. 898-906.
- Dahms, B.B., T. Boyd, and R.W. Redline, Severe perinatal liver disease associated with fetal thrombotic vasculopathy.Pediatr Dev Pathol, 2002. 5(1): p. 80-5.
- Ernst, L.M., A.B. Grossman, and E.D. Ruchelli, Familial perinatal liver disease and fetal thrombotic vasculopathy.Pediatr Dev Pathol, 2008. 11(2): p. 160-3.
- Redline, R.W., Severe fetal placental vascular lesions in term infants with neurologic impairment.Am J Obstet Gynecol, 2005. 192(2): p. 452-7.
- Parast, M.M., C.P. Crum, and T.K. Boyd, Placental histologic criteria for umbilical blood flow restriction in unexplained stillbirth.Hum Pathol, 2008. 39(6): p. 948-53.
- Tantbirojn, P., et al., Gross abnormalities of the umbilical cord: related placental histology and clinical significance.Placenta, 2009. 30(12): p. 1083-8.
- Saleemuddin, A., et al., Obstetric and perinatal complications in placentas with fetal thrombotic vasculopathy.Pediatr Dev Pathol, 2010. 13(6): p. 459-64.
- Ryan, W.D., et al., Placental histologic criteria for diagnosis of cord accident: sensitivity and specificity.Pediatr Dev Pathol, 2012. 15(4): p. 275-80.
- Redline, R.W. and A. Pappin, Fetal thrombotic vasculopathy: the clinical significance of extensive avascular villi.Hum Pathol, 1995. 26(1): p. 80-5.
