Introduction
For the baby to thrive, the mother must provide the placenta with a large blood flow from her uterine circulation. The increase in uterine blood flow to the placenta has been estimated at 8 to 10 times baseline flow. The body can increase blood flow to a local tissue by relaxing the muscles around arterioles (small arteries), for example when an inflamed area becomes red or when a athlete needs to shunt more blood to active muscles. This strategy works because resistance to blood flow is lowered in this segment of the circulation compared to other areas. Unfortunately this usual strategy of widening arterioles is not able to provide enough increased blood flow for the placenta. A unique strategy is required that is specific for pregnancy and the placenta. This process can go wrong. If it does the fetus may not get enough nutrients or oxygen. The mother may develop preeclampsia. The placenta must adapt to decreased blood flow, and may suffer placenta infarctions and even premature placental separation. This chapter will focus on the normal and pathologic changes in these uterine vessels.
My understanding of the basic physics of blood flow
The basic physical principle is that blood flow is directly related to blood pressure (push harder and more will flow) and indirectly to resistance (the less force needed, the more blood will flow). The resistance is fundamentally due to friction against the blood vessel wall, the wider and shorter the blood vessel, the less the resistance to flow. Poiseuille’s equation relates the flow of a Newtonian (non-turbulent) fluid in a tube, and although blood flow in a vessel may not meet all the conditions of the equation, it provides a good approximation for understanding the basic parameters governing blood flow.
| Q = | ΔP π r4
___________ 8 L η |
Q is the blood flow. ΔP is the pressure change, which in the closed circulation is the difference between the arterial and the venous pressure. R is the radius of the blood vessel. The wider the vessel, the less of the flowing blood is rubbing against the vessel wall. The volume passing through is related to the area of the cross section πr2, while the wall circumference increases linearly πr. This relationship will apply for the whole length of the blood flow (L). The longer the vessel, the more resistance. The more viscous the blood (η), the more resistance. If the pressure gradient, and the length of the vessel and viscosity are all kept constant, increasing the radius of the vessel by 2 should increase the flow by a factor of 16!
A larger radius will also have an effect on the flow velocity. For the same pressure change, if more volume is being moved, the velocity will slow down because of conservation of momentum. My understanding is that this is a closed circuit, and the mass is at a certain velocity in the lower radial artery, as the vessel widens, the volume becomes larger, and in order to keep the same mass moving through the system, this mass must be moving at a slower speed. This slower flow will aid gas and nutrient transfer between mother and fetus.
What is the normal circulation of the uterus?
The standard picture of the body’s circulation is a symmetrical pattern of dichotomous branching in which each vessel branches into to two smaller vessels until the capillaries are reached, and then the process reverses going with dichotomous fusing of veins into progressively larger vessels. This fractal pattern holds more or less in most organs, except for collateral vessels that branch across the pattern to provide an alternative route of blood.
The arterial pattern in the uterus follows the expected pattern from the main uterine arteries that branch dichotomously through the outer connective tissue (serosa) and the muscular body (myometrium). There are radial arteries that run perpendicularly across the myometrium, somewhat anomalously to the usual pattern, that are critical to the development of the placental circulation, especially in the endometrium. This endometrial circulation because of menstrual shedding and pregnancy has some unique attributes.
My understanding of the uterine circulation was based on reading the studies of the endometrial circulation in the monkey published by Elizabeth Ramsey. She and other have cited critical earlier studies, but her monograph on the topic is a lucid and insightful summary. Her paper of her injection studies in the original Carnegie Institute Contributions to Embryology paper shows the frustrations and painstaking effort of those studies1. Her monograph on the subject with Martin Donner “Placental Vasculature and Circulation” details her impressive radiologic injections as well as the anatomic injection studies 2.
I will briefly relate my working understanding of the monograph. Her work was done using monkeys as subjects. They differ from humans in a few details, such as not developing swollen decidual cells in the endometrium and forming a secondary placental lobe on the opposite wall of the uterus. Perhaps most importantly, the human placenta over the pregnancy expands the base of involved arteries tapped for the placenta beyond those of the implantation site, unlike other primates. The rhesus monkey typically utilizes 8 to 12 arteries compared to 120 to 320 in humans. (I have trouble understanding this because studies of essentially “cotyledons” of spiral artery flow into the placenta number 30-40. This requires some kind of consolidation of groups of spiral arteries.) Other differences include the thinning of the human endometrium and the marked expansion of blood flow beyond that in the primate. At the end of pregnancy the uterine vessels that pre-pregnancy carried a few milliliters of blood per minute, carry approximately 600 ml/min.
Anatomic injection studies show that the radial arteries are continuous with the spiral arteries of the endometrium, and there is in the base of the endometrium basal arties that extend horizontally from the base of the radial arteries and are not modified by pregnancy. The spiral arteries undergo progressive dilatation throughout pregnancy, a process that extends into the myometrium (Fig 1).
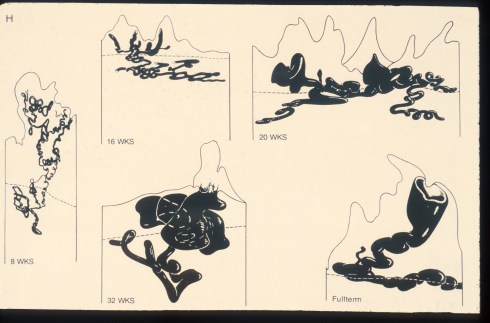
Fig 1: This is an illustration from Dr. Ramsey of the India ink injections of the monkey showing the progressive dilatation of the spiral arteries through gestation.
The average diameter of the spiral arteries before pregnancy is 200 microns, and this enlarges to 500 microns at term. The basal arteries in the endometrium remain at an average diameter of 180 microns3.
The radiologic injection studies demonstrated that the spiral arteries filled the intervillous space intermittently (Fig 2).
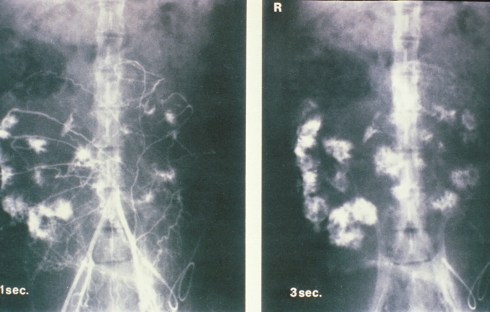
Fig 2: This is an illustration from Drs. Ramsey and Donner’s monograph of radiologic injection into the uterine circulation of the pregnancy monkey. The entire placenta is not circulated but rather separate spiral arteries in this one injection sequence.
Even in mid-pregnancy , the weak uterine contractions inhibited uterine blood flow. During labor contractions, blood remained in the intervillous space providing some gas exchange even during the cessation of flow due to uterine contraction.
Other prominent students of the utero-placental anatomy include Maurice Panigel4, and the Hamilton and Boyd monograph on the human placenta5, among many others. Some of the history of our understanding of this anatomy is cited in Ramsey and Donor monograph and in a 2006 summary of our knowledge of the spiral arteries by Pijnenborg and colleagues6.
How does the body widen the uterine vessels?
There are two potential ways that a blood vessel could widen. First it could simply grow bigger by proliferating the muscle and connective tissue cells of the wall. This is the way vessels normally respond to increased blood flow as organs grow. The increased flow increases pushes the vessel wall outward. This wall tension is counteracted by cell proliferation increasing the diameter and thickness of the vessel to a larger sustainable size for the flow and pressure.
The second possible way that a blood vessel may widen is that a weakened wall simply expands from the wall tension until a new equilibrium is reached, or the wall ruptures. The latter is occurs often with devastating effect in the focal wall weakness that produces an aneurysm. In pregnancy, the arterial vessels supplying the placenta are widened by destruction of the vascular media in a circumferential process. The possibility of progressive aneurysmal dilatation as an occasional cause of thrombosis or of vascular rupture and hemorrhage cannot be excluded. The coordination of the loss of the media of the vessel involves extravillous trophoblast invasion outside the vessels as well as intravascular trophoblast undermining of endothelial cells. Initially, the vascular lumen may be plugged with trophoblast. At the conclusion of the remodeling the vessel is widened with its wall replaced by fibrinoid and intravascular cytotrophoblast. Numerous biochemical communications are involved including signals from endothelial cells and inflammatory cells. A coordinated quantitative hypothesis of these signals in space with the integration of cell responses does not yet exist6.
Can the pathologist see the trophoblast remodeling in the spiral arteries?
The pathologist may observe spiral arteries in curettage samples from spontaneous abortions as well as in the decidualized endometrium attached to the delivered placenta and membranes. These are limited samples either because they are not oriented from the curettage or partial vessels from the separation of the decidua. Less frequently, direct, oriented visualization of deeper spiral and radial arteries is possible from uteri of gravid hysterectomies or autopsies. These chance observations of spiral arteries, as well human placental bed biopsies taken in the context of research, have informed the development of pathologic observations and diagnoses.
I intend to ask colleagues to discuss the large volume of papers on the subject of placental bed biopsies. Much of how we interpret changes in other decidual and myometrial samples is informed by these studies.
The pathologist can evaluate the adequacy of utero-placental flow by the adaptations of placental villi, a topic that will be considered in a later section. The role of utero placental perfusion in intrauterine growth restriction is potentially another measure of trophoblast vascular remodeling that will also be discussed in a subsequent section.
What do the spiral arteries demonstrate in early pregnancy curettage?
Pathologists see the range of changes described by more systematic research of the trophoblast remodeling of the spiral arteries. The infiltration of the decidua by giant trophoblast cells is often a prominent feature. Plugging of spiral arteries can be seen. Endovascular invasion of spiral arteries may be present (Figs 3,4).
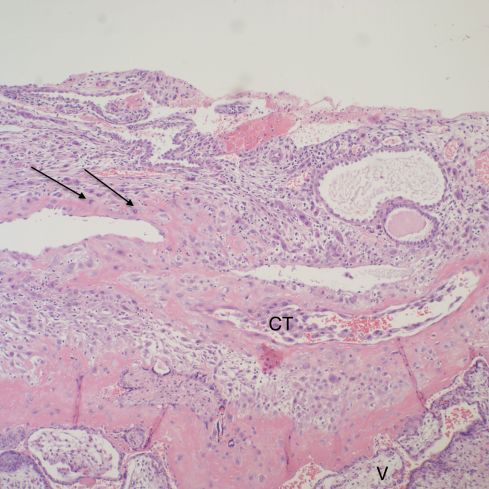
Fig 3: The top portion of decidua demonstrates embedded endometrial glands with plugged mucus in the lumen. Beneath them are two profiles of a remodeled spiral artery. On the left, the arrows point to cytotrophoblast replacing the muscularis. To the right the endothelial lining has been replaced by cytotrophoblast (CT). There are cytotrophoblast, including multinucleated cells surrounding the vessels. At the bottom is the basal cytotrophoblast/fibrinoid layer, and the immature villi can be seen below that band (V). H&E, 10x
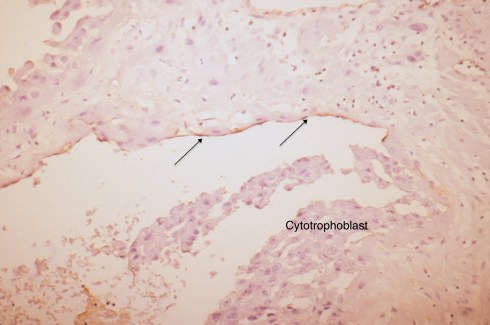
Fig 4: The arrows show the brown immuno-staining that identifies endothelium lining a vascular space, presumably a spiral artery devoid of muscularis. The large irregular cells in the lumen are typical cytotrophblast within the vessel that are undermining the endothelium, and will replace it. CD31
The findings are a mix of necrotic decidua capsularis, which occurs over the implantation, the decidual basalis that is beneath the placenta and the parietal decidua beneath the membranes. Elective abortion curettage has not been available to me for examination, so there is no normal gestation matched control tissue. There is decidua from medical induced abortions for lethal anomalies, but I have not systematically compared them to examples with known causes of fetal loss. I attempted a comparison of fetal loss curettage findings in diabetic pregnancies with non-diabetic, and did not find significant differences7.
What does study of the whole gravid uterus demonstrate about the spiral arteries?
The uterus may be surgically removed following delivery for uncontrollable uterine bleeding, usually due to some form of placenta accreta, but occasionally for uterine atony. In the latter, the uterus fails to contract despite therapeutic injection of uterine stimulants and physical massage. Less commonly, the uterus is included in the autopsy of a maternal death. Forensic pathologists may also examine gravid uteruses from causes of death not directly related to obstetrical disease such a trauma or homicide. There have been some publications of systematic reviews of whole gravid uteri 8and of maternal autopsies with toxemia 9.
I regret not having explored and recorded more of the vascular anatomy in the uteri that I examined. The focus was on the diagnostic features of accreta, or possible causes of atony. One incidental observation that I could not explain in a uterus removed for placenta accreta was severe chronic inflammation in a deep myometrial artery in which a portion of the artery was necrotic suggesting trophoblastic remodeling (Figs 5a, 5b).
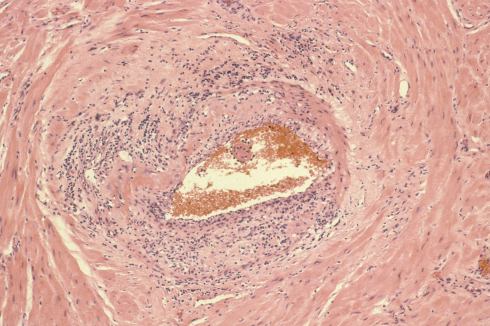
Fig 5a: The artery in the center is surrounded by large gravid myometrial myocytes. Its wall on the right is intact, but the left, top and bottom shows loss of medial smooth muscle, numerous inflammatory cells, and smudged eosinophilic material in the media. H&E 10x
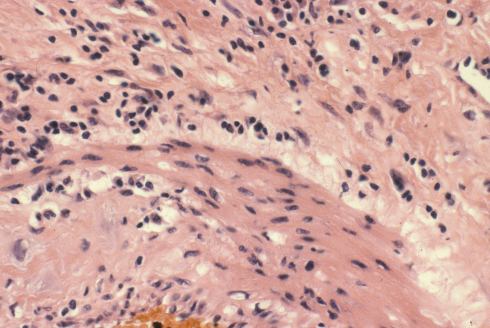
Fig 5b: This image is a higher magnification of the above artery at the junction of intact and damaged media. The inflammatory cells appear to include neutrophils, lymphocytes and even plasma cells. H&E 40x
What do the delivered placenta and membranes demonstrate about the spiral arteries?
The placenta and membranes separate in the plane of the decidua. Fragments of the superficial spiral arteries are included within the base of the placenta and the decidua of the membranes.
Un-remodeled muscular spiral arteries are usually found in the membranes. They may be difficult to distinguish from the deeper, smaller radial arteries. The most striking pathologic lesion of these spiral arteries is acute atherosis, which will be discussed below. The arteries may demonstrate numerous mononuclear cells in or around the wall of the spiral arteries, or even eosinophils (Figs 6a, 6b, 7).
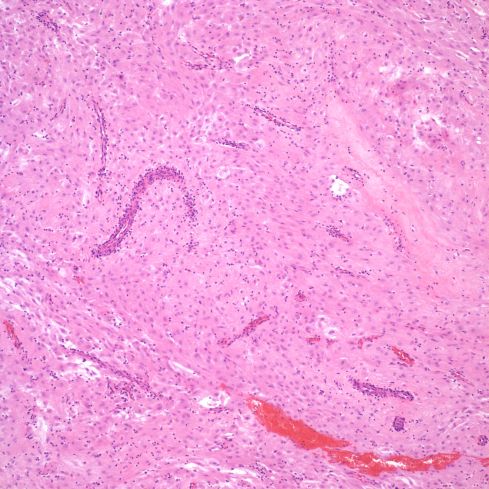
Fig 6a: This is a low power image of a spiral artery cut in the parallel plane of the delivered parietal decidua. Various profiles of a thin walled spiral artery can be seen, and many are surrounded by inflammatory cells. H&E 10x
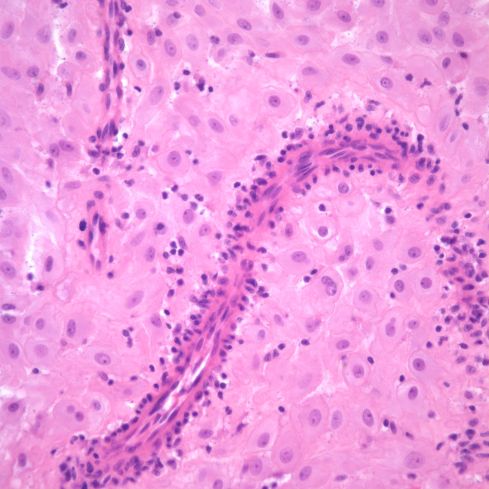
Fig 6b: A higher power of one of the spiral arteries in the previous section demonstrates the lymphocytic appearance of the inflammation. H&E 40x
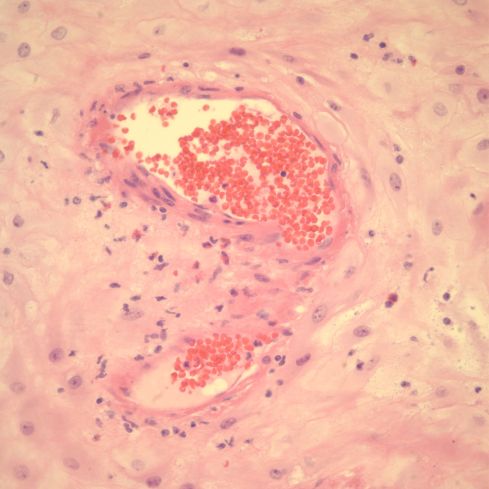
Fig 7: This spiral artery in the parietal decidua shows eosinophils surrounding a thin walled spiral artery. H&E, 40x
Thin walled vessels may show thrombi although many of these may be in venules (Figs 8, 9).
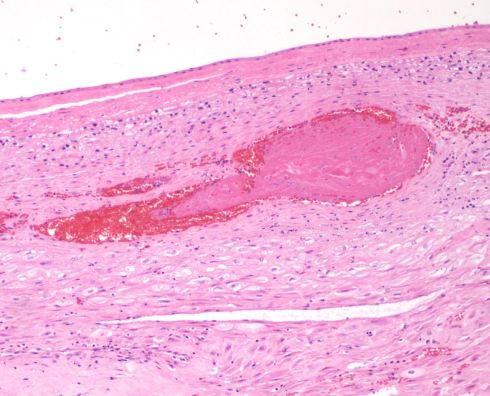
Fig 8: This sample of the parietal membranes is a transverse section with the amnion at the top. There is thrombus is a thin walled vessel with necrosis of the media. H&E 10x
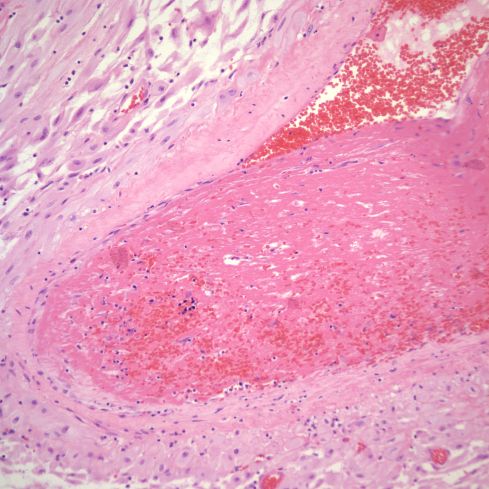
Fig 9: This is another example of a thrombus in the parietal decidua showing a thrombus with medial necrosis of the vessel, likely a spiral artery. There is very early organization of the thrombus. H&E 20x
The significance of these later two findings is unknown.
The spiral arteries of the membranes even in term pregnancies may show changes of trophoblastic remodeling of spiral arteries. The chorionic epithelium of the membrane may appear to have loosened and descended into the trophoblast (Figs 10a, 10b, 11,12).
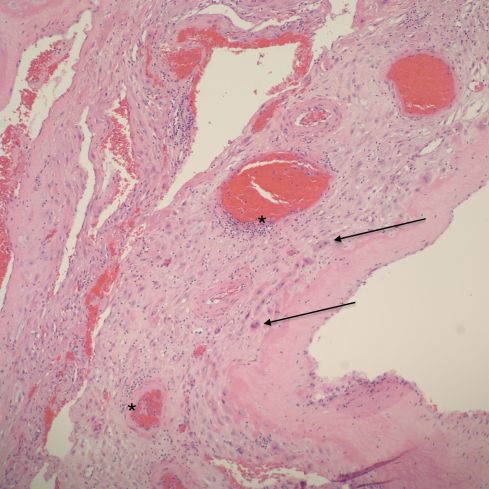
Fig 10a: This sample of parietal membrane demonstrates loss of the intact cytotrophoblast layer with many independent often multinucleated trophoblast cells (arrows). There are multiple profiles of a spiral artery. Some are normal, and others show dilatation, thinning and necrosis of the media and focal inflammation (*). H&E 10x
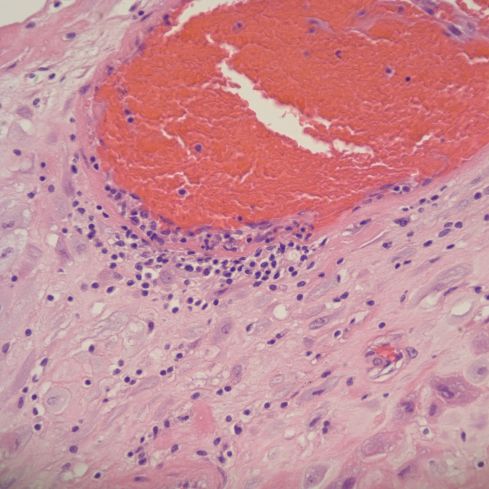
Fig 10b: This higher power of the area of the “*” in the previous figure shows the eosinophilic necrosis of the media, and the mononuclear inflammation. The red cells appear distorted suggesting stasis. H&E 40x
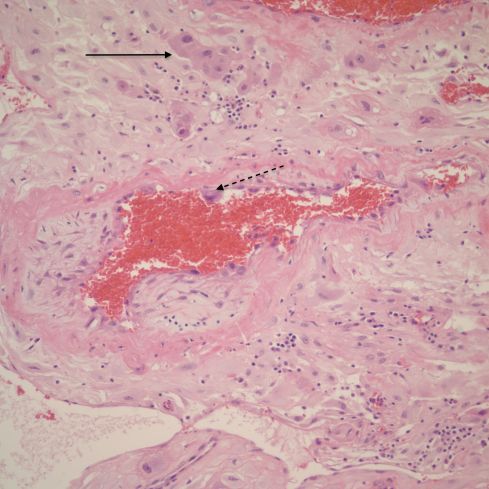
Fig 11: This spiral artery in the parietal decidua demonstrates destruction of the media partially showing early organization (scar formation) and a lining of cytotrophoblastic cells (dotted arrow). Loss cytotrophoblast are present in the decidua (arrow). H&E, 20x
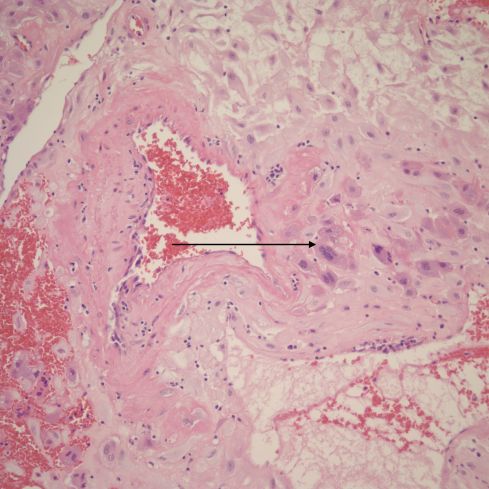
Fig 12: This is another example of a remodeling spiral artery in the parietal decidua. The arrow points to a multinucleate trophoblast. H&E, 20x
Such often multinucleated trophoblast may be migrating from the margin of the placenta (Fig 13).
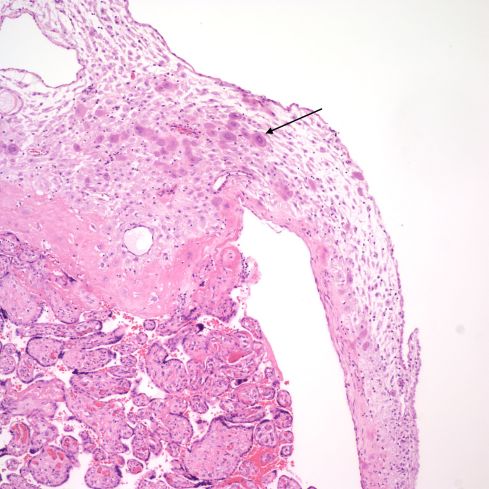
Fig 13: The placental villi are at the bottom, and the basal decidua is at the top. There are multinucleate trophoblast cells that extend into the marginal decidua (arrow). H&E 10x
This has no known significance. This will be discussed in more detail in relationship to acute atherosis.
The spiral arteries may have a thickened media, which has been considered one form of decidual vasculopathy. I do not know of specific, quantified diagnostic criteria correlated with clinical income. Spiral arteries may appear thickened because of cell swelling, interstitial edema and interstitial fibrinoid material, as well as by smooth muscle hyperplasia10. The normal thickness may depend on gestation as well as nearness to areas of invasive cytotrophoblast (Figs 14-18).
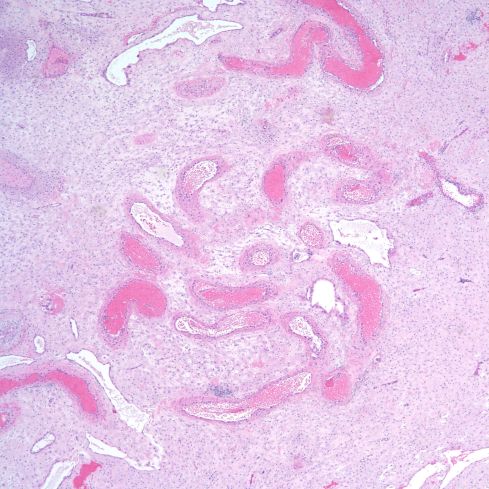
Fig 14: Low power of a spiral artery in the parallel plane of the parietal decidua showing a media of sparse myocytes of which the thickness appears to vary because of oblique cutting of the vessel. The mother suffered from diabetes and chronic hypertension. H&E 4x
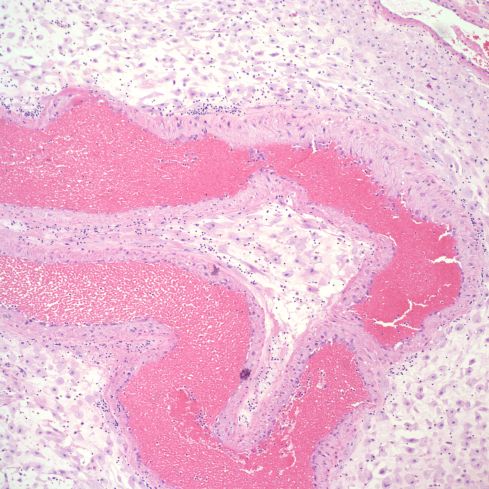
Fig 15: A higher power magnification of the same artery as in Fig 14 showing the myocytes more clearly. H&E 10x
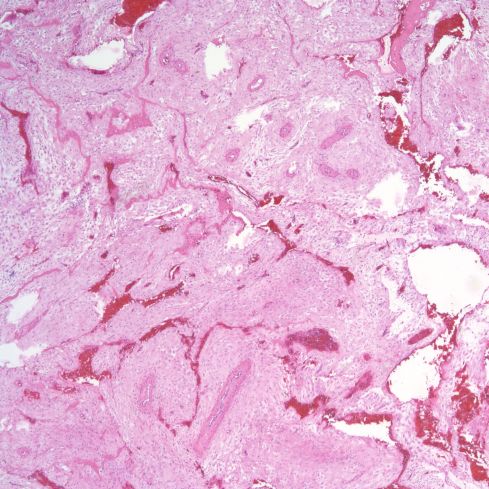
Fig 16: This is an image similar to Fig 14 from a normal term delivery in which the spiral artery appears to have a thicker media. H&E 4x
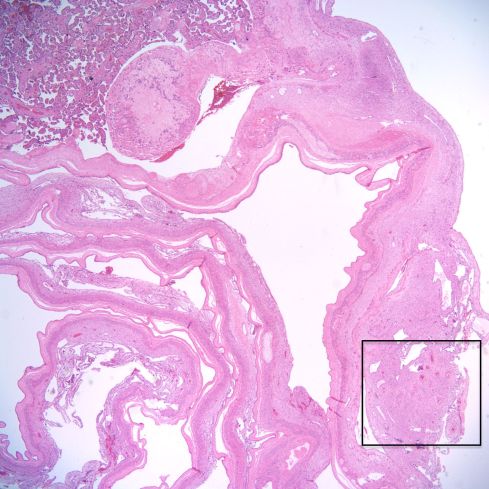
Fig 17: This is a low power image of the membranes arising from the margin of the placenta. The rectangle shows a spiral artery in the membranes adjacent to the margin of the placenta. H&E 2x
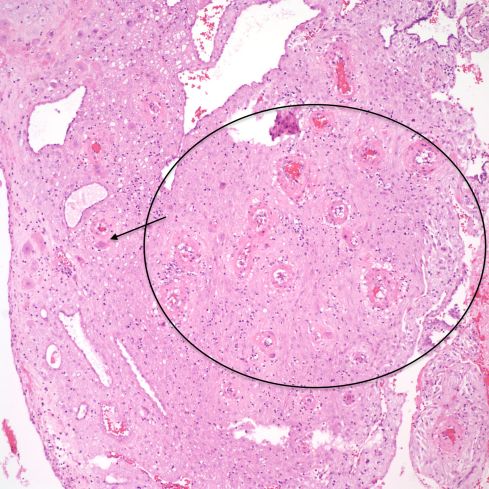
Fig 18: This higher power of the above sample shows that the apparent thickened spiral artery is due to swelling from trophoblast-induced damage (circle). The trophoblast can be seen in the margin (arrow) H&E 10x
The spiral arteries at the base of the placenta have been transformed by trophoblast and often are only multiple profiles of a tortuous vessel shell lined by a thick hylaine material, usually with embedded cytotrophoblast cells. As in the decidua of the membranes, there may be basal arteries. Untransformed spiral arteries may be present and have been considered a possible marker for under-remodeling of spiral arteries.
A perhaps understudied aspect of the spiral arteries in the basal decidua is their appearance when they underlie lesions of placental infarction, retroplacental hematoma, subchorionic thrombohematoma, and infarction hematoma. Those underlying infarctions may be distended with blood and thrombus (Figs 19-24).
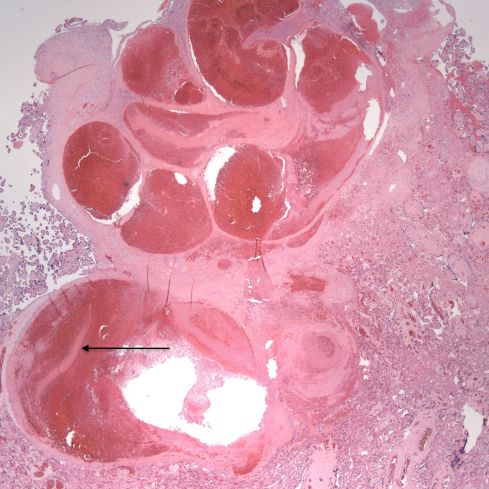
Fig 19: The large, blood filled convoluted spiral artery at the base of the placenta shows lamina of fibrin. The infant was liveborn at 26 weeks of gestation, but was severely growth restricted. The mother had type 1 diabetes and chronic hypertension. The placenta had multiple infarctions. (H&E, 2x)
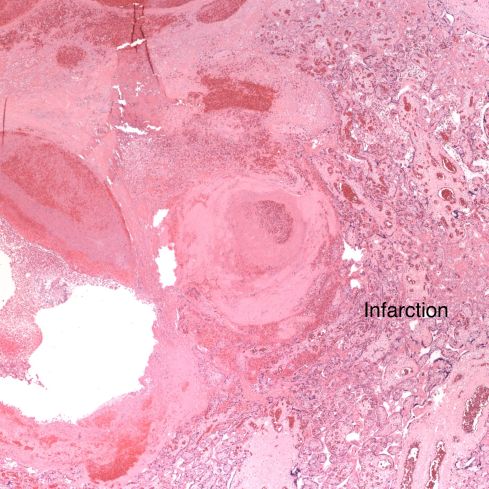
Fig 20: This view of the same spiral artery shows is proximity to a recent placental infarction. The presence of a thrombus in the vessel suggests that the infarction may have been caused by this very superficial thrombus, but there is no obvious clue as to how it formed. (H&E, 2x)
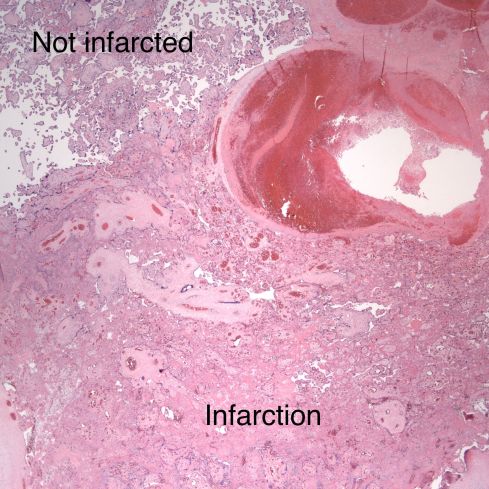
Fig 21: Another basal spiral artery from the same patient as in Fig 19 showing fibrin lamina and underlying a very acute placental infarction. (H&E, 4x)
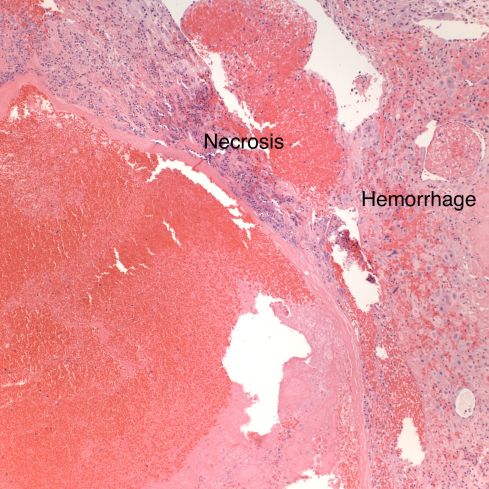
Fig 22: In another area of the above vessel the infarction involves the basal decidua (“necrosis”) as well the villi. Surrounding viable appearing decidua shows acute hemorrhage between the decidual cells. A gross retroplacental hematoma was not present. (H&E, 10x)
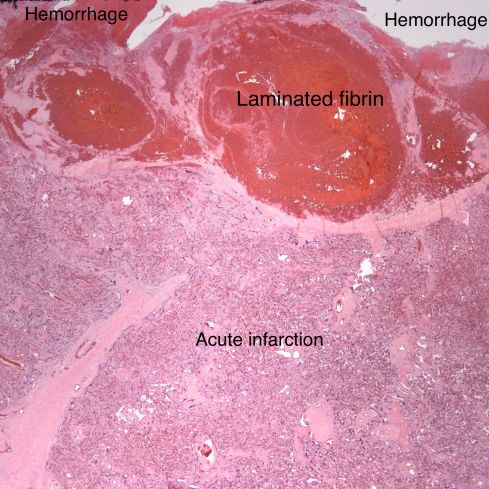
Fig 23: The base of the placenta demonstrates a dilated spiral artery with laminated fibrin and an acute overlying placental infarction. Around the spiral artery hemorrhages can be seen in the basal decidua. The placenta had multiple infarctions and this small retroplacental hematoma. The infant was small for gestation with Apgar scores of 7 &8. (H&E, 2x)
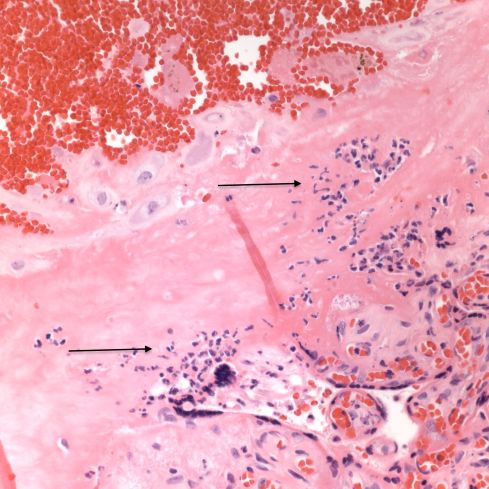
Fig 24: A high power over the base of the placenta above the retroplacental hematoma demonstrating an influx of neutrophils, a typical inflammatory response to a retroplacental hematoma (arrows). (H&E, 40x)
In older infarctions, the spiral arteries may demonstrate sparse red cells with loss of hemoglobin consistent with vascular stasis or with partial mural thrombi (Figs 25-29).
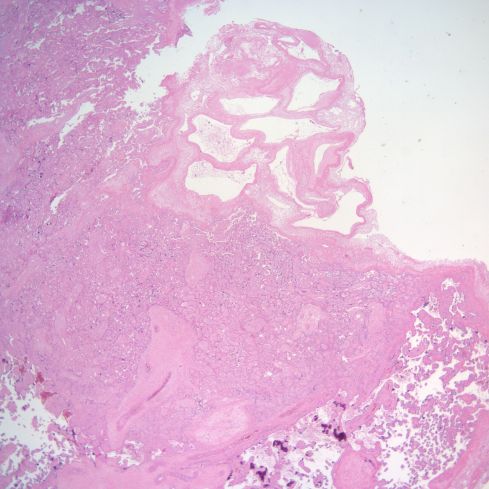
Fig 25: The infarction shows the compacted pale villi in the lower portion of the image. Above the infarction a complex spiral artery profile shows lumen empty except for some faded blood cells. (H&E, 2x)
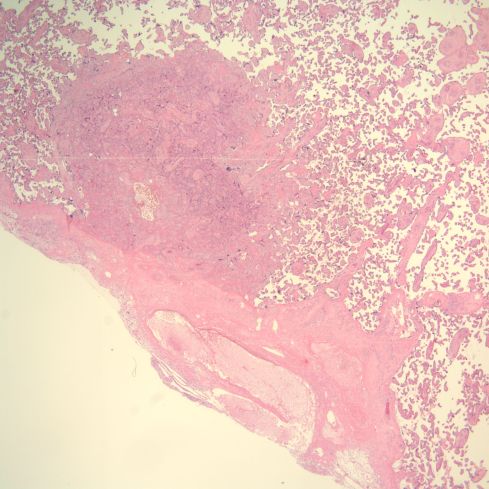
Fig 26: This is another infarction, oriented above the base of the placenta, which runs across the lower left corner of the image. In the lower left of this diagonal can be seen a dilated spiral artery that appears empty except for faded blood cells. There is no thrombus at this level of the superficial decidua, although the infarction is evidence of vascular occlusion in the spiral artery. (H&E, 2x)
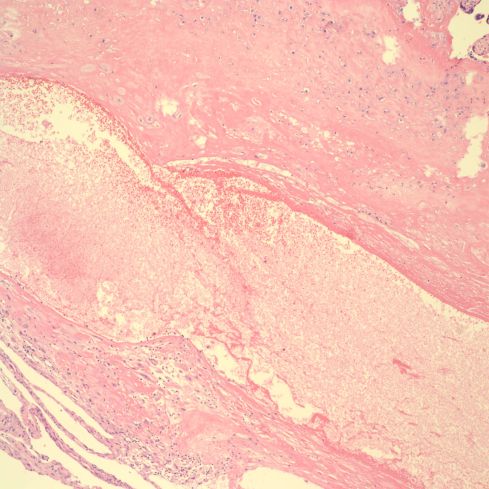
Fig 27: This is a higher power image of the above artery showing the thin wall without a definable media. (H&E, 10x)
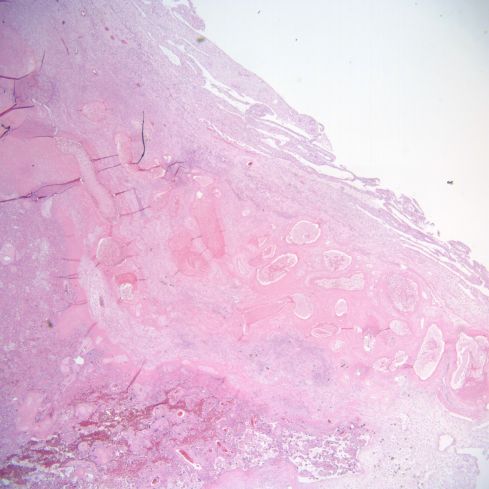
Fig 28: This is similar to the above two cases. The infarction to the left with the base of the placenta shows above it in the image. The base demonstrates a long, complex set of cross sections of a spiral artery showing old, pale red cells. (H&E, 2x)
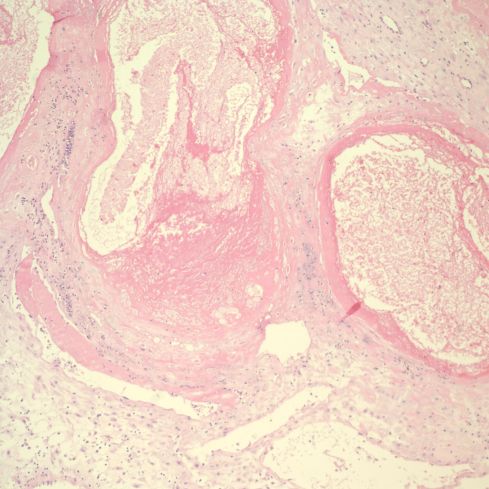
Fig 29: The vascular profile in the right corner of Fig 28 demonstrates some non-occlusive mural thrombus in this higher power image. (H&E,10x)
The implication is that the occlusion of the vessel, mostly likely from thrombus, occurred proximal to the portion of spiral artery in the most superficial dedicua. In retroplacental hematoma, vessels may demonstrate rupture and hemorrhage into the underlying decidua. In infarction hematoma, a spiral artery may appear to rupture into the intervillous space. In most cases of these complications, no spiral artery is identified, likely because it has not been sampled, or is no longer attached to the placenta.
The lining of base of the placenta on the intervillous surface appears to have an endothelial phenotype, as do vessels that are likely veins. The arteries maintain a trophoblast phenotype, but more systematic studies may show less of a dichotomy (Figs 30,31).
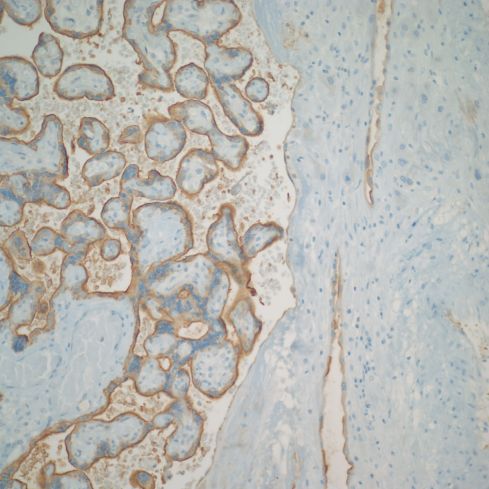
Fig 30: This normal mature placenta demonstrates villi with brown staining from an antibody to placental alkaline phosphatase (PLAP) present on the surface of the villous trophoblast. The basal decidua runs along the right of the image. The spiral arteries are also lined by brown stained cells indicating that the lining cells are trophoblastic. (PLAP, 20x)
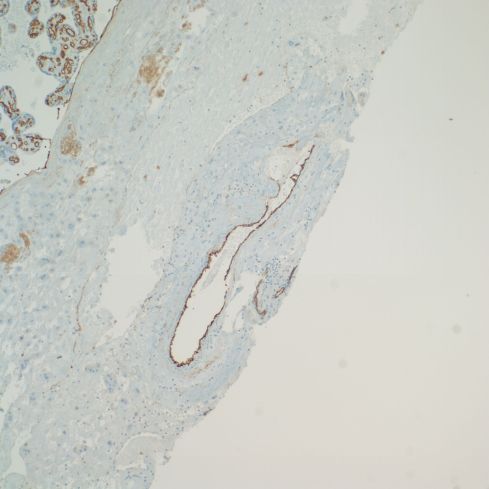
Fig 31: This is the same placenta showing brown immunostaining with an antibody to platelet endothelial cell adhesion molecule (CD31) that stains endothelial cells in the fetal vessels in the villi, and the lining of a vessel in the decidua, which may be a large vein, but a deeper portion of a spiral artery cannot be excluded. (CD31, 10x)
Is there any clinical diagnostic value in examining the spiral arteries?
The goal of the placental pathologist is to find lesions that either explain the outcome of the current pregnancy, or even better predict the problems of the next pregnancy or of possible subsequent maternal disease. Subsequent sections will discuss toxemia (and the varied terms for this disease(s)) and discuss the causes of fetal growth restriction. However, both of these clinical complications may be related to a failure of trophoblast remodeling of the spiral arteries. Hence, logically, pathological examination of these arteries may clarify mechanisms of these complications.
The origin of the failed spiral artery remodeling is still unresolved. In a 2010 review article that presents numerous control factors that may be involved in trophoblast remodeling, the authors lament that the mechanisms that these interdependent mechanisms have primarily been considered in isolation11. They conclude “It is only when we have a clearer picture of the events occurring in a normal pregnancy that we can begin to determine which of these are compromised in pregnancies complicated by disorders such as pre-eclampsia.” In the subsequent almost one decade, there have been studies elucidating the roles of natural killer cells, local hemodynamics, and numerous biological signals including the effects of certain microRNAs on tissue culture models. I still cannot see a comprehensive quantitative model of the process of trophoblast remodeling. I think achieving that model would be a breakthrough that would have implications for more that preeclampsia. Yet, to understand the origin of preeclampsia, at least to me, looking for upstream events that differentiate first pregnancies from subsequent pregnancies might provide some therapeutic leverage even if the understanding of trophoblast modeling is incomplete.
The most clinically salient clue as to the origins of toxemia is that it predominates in the mothers’ first pregnancy. This suggests that looking at key differences between the first and subsequent pregnancies might point to the initiating pathogenesis. One clue that simple early hormonal differences could play a role is provided by primate studies that administer first trimester estrogen to interfere with trophoblast remodeling of spiral arteries12. A delay in starting the remodeling might be enough to subvert it. I do not know if first pregnancies are slower to elevate progesterone over estrogen, but hormonal differences could be important. Another upstream observation is a reported decrease in hyperglycoslated HCG in toxemia 13. The suggestion is that hyperglycoslated HCG prevents TGF beta induced cytotrophoblast apoptosis and promotes invasion. Finding the upstream events initiating toxemia is a reasonable approach until we can comprehend the regulation of the whole process.
This thought allows re-phasing the original question as “can the examination of the spiral artery demonstrate early mechanisms of failed trophoblast remodeling. One potential study is to compare the spiral artery findings in first trimester primigravida pregnancies to those in multiparous mothers, and look for statistical differences. Most of the specimens that I have seen are from spontaneous abortions that may not represent normal trophoblast remodeling, but this approach could be productive if such specimens were available. Another approach is to examine delivered placentas to find evidence of difference between primigravida and multigravida placentas, or between patients with different clinical patterns of toxemia. Stanek reported observations of the placenta and membranes after delivery to identify “shallow implantation” which I interpret to be the same as the incomplete trophoblast remodeling14. These features included those of spiral arteries including hypertrophic arteriopathy and atherosis, as well maternal floor trophoblastic giant cells and excessive numbers of extravillous trophoblast. These features distinguished normal from preeclamptic (toxemic) pregnancies, but not early from late onset disease. The implication is that these clinically different patterns do not differ histologically on key features of the spiral artery changes. This study has large numbers of cases recorded by an experienced pathologist but the conclusions would be strengthened if tested in a blind prospective study with concordance of diagnoses among a group of pathologists.
Another effect of toxemia is an increased prevalence of placental infarctions and retroplacental hematomas including those producing clinical abruption of the placenta. While these lesions will be considered in other sections and chapters, looking at the spiral arteries underlying these lesions may be of value. The underlying causes of infarction and abruption in toxemia may be the same or different from that occurring in other clinical disease. These differences could have predictive value for future pregnancies. I am unaware of a successful attempt to make these distinctions, but there is more work to be done.
What is acute atherosis?
The one distinct anatomic diagnosis of the spiral arteries in pregnancy is acute atherosis. Its clinical significance is still uncertain. This lesion is part of a triad that is accepted as decidual vasculopathy15. Two of the components I will lump together as acute atherosis, the shiny eosinophilic fibrinoid necrosis of the arterial wall and the accumulation of lipid filled macrophages in the fibrinoid (Fig 32,33).
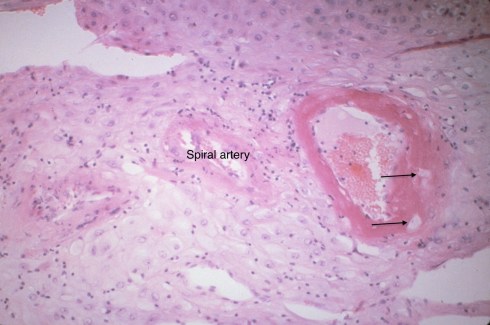
Fig 32: The specimen is from the decidua attached to the fetal membranes in a mother with toxemia. There are three cross sections through a spiral artery. The cross section on the right shows a dilated portion with loss of muscular wall. Where the muscular wall would have been there is a thick, pink (eosinophilic), smudged appearance. This is the fibrinoid necrosis. Within this structure, the arrows point to foamy clear macrophages. There are also scattered mononuclear inflammatory cells.
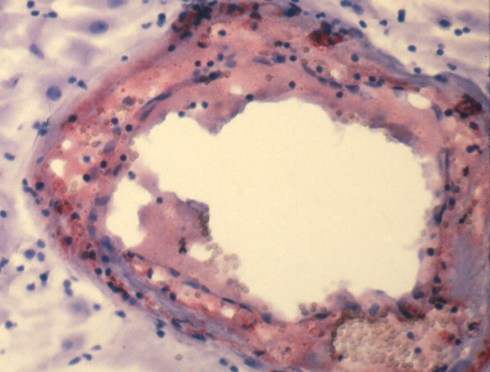
Fig 33: A high power frozen section of a spiral artery from the same placenta in Fig 32 stained with Oil Red O to detect fat. Small globules of the fat can be seen in the fibrinoid of the vessel.
Acute atherosis is a lesion of untransformed spiral arteries. Fibrinoid is a pathologic term that simply implies that under the microscope, the pink amorphous material looks like fibrin that has deposited, but it does not imply actual thrombus formation. There is still some debate about the nature of the pink material. The necrosis of the smooth muscle, increased endothelial permeability to plasma proteins and complement deposition has all been invoked as contributors to the appearance. The lipid cells have been identified as macrophages based on CD68, a lysosomal marker. The bulk of the lipid presumably comes from transudation of serum proteins, analogously to atherosclerosis. The role of inflammatory cells, which may be in or around the vessel wall, is unknown. Often there are no inflammatory cells in direct proximity to the lesion. The lesion is often segmental involving only some histologic profiles of the artery. The vessel is usually dilated.
As far as can be discovered, including in Sheehan and Lynch’s monograph of 677 maternal autopsies of mothers with toxemia, acute atherosis is restricted to uterine spiral arteries9. (Sheehan and Lynch did find infrequent infiltration of arterioles with diffuse lipid with fat stains in the kidney and liver, but without vascular necrosis.) Any theory of the pathogenesis of acute atherosis would have to explain not only the anatomic restriction to spiral arteries but other observations. First, why is the lesion is segmental in one artery, but also why does it only involve some of the arteries in the same decidual area. Second, why do only a minority of women with toxemia demonstrate the lesion? Third, why is the lesion present in some patients without clinical toxemia. Spiral arteries are unique not just because of acute atherosis but also that during the menstrual cycle they respond to hormonal stimuli, and finally shed to regrow with the next cycle. The decidual environment is also unique and of perhaps relevance, decidual cells appear to produce renin, the initiating enzyme in the vasoconstrictive angiotensin system.
An important attempt to explain acute atherosis was published in a review article by Carlos Labarrere who was working with Paige Faulk at the Center for Reproduction and Transplantation Immunology at Methodist Hospital in Indianapolis IN, when I met him16. He reviews the work of the English group on placental bed biopsies and the hypothesis that PIH, as well as unexplained IUGR, is associated with placental arteries that have not undergone trophoblast invasion. He also considers the studies of acute atherosis observed in diabetes, hypertension without PIH, unexplained IUGR and autoimmune disease. He points out fluorescent studies of atherosis show large IgM and C3, and some C1q. He says similar deposits are seen in rejection lesions with lipid deposition. In describing rejection, he points out lipid starts in myointimal cells and is part of insudation from damaged endothelium. The finding of IGM and complement in acute atherosis is consistent with an immune complex disorder perhaps related to a fetal antigen. The insudation of lipid does not appear to be simply leakage of serum proteins through the damaged endothelium. Immunofluorescent studies of acute atherosis did not localize albumin or transferrin in the vessels. Dr. Labarrere argues that with an abnormal immune response, fetal antigens in excess would generate IgM instead of a protective IgG response. He cites some serum evidence of immune complex disease in PIH that I have not reviewed. Dr. Labarrere’s review was published in 1988, yet many of the same questions remain today.
A note about his cited immuno-fluorescent study published in 198117. The incidence of acute atherosis in patients with preeclampsia was 53%, which is high. They also found the lesion in patients with normotensive insulin dependent diabetes and with stable chronic hypertension. This study was done on curettage specimens based on palpating the location of the placenta before delivery, and then scraping that area after delivery. The diagnosis of acute atherosis was made on frozen samples. They recognized that trophoblast transformed vessels could also show lipid in from degenerated cytotrophoblast in the walls of these vessels. Controls opposite the placental bed and in normal pregnancies did not show immunostaining. Since insulin dependent diabetes could be associated with endothelial damage, the lesions resemblance to that seen in preeclampsia is not surprising. I have not seen the lesion in fetal membranes in this context. However, more aggressive management of serum glucose in these patients may have decreased the underlying vascular pathology. The cases of acute atherosis in some normotensive patients with systemic lupus erythematosis would support an immune complex derivation of the atherosis. I wonder if redoing of immuno-localization studies using antibodies to C4d might not help clarify whether immune complexes are involved in the pathogenesis. Using arteries from the fetal membranes would reduce possible diagnostic confounding with trophoblast remodeled vessels.
Why is acute atherosis only present in some women with toxemia?
Acute atherosis occurs in only a minority of patients with toxemia. A potential explanation for this observation is that the prevalence of a focal lesion is being underestimated as a result of limited sampling. Most of the untransformed spiral arteries seen by the pathologist are in the decidua attached to the fetal membranes. Pathology assistant Yasser Daoud and I presented a small study at the first Mid America Placenta Study Group meeting in Toronto in 1998 that tried to answer this problem. We sampled the fetal membranes by taking a 1-2 cm roll of membrane from the rupture site to the placental margin. Two cross sections of a single membrane roll were included in a histology-processing cassette. In 25 cases with a clinical diagnosis of pregnancy-induced hypertension, three additional membrane rolls were processed. Nineteen patients had no atherosis on any of 4 slides, 3 had atherosis on one slide, and three had atherosis on two slides. In two cases, the first sample had the atherosis. Further sections diagnosed four additional lesions. (Interestingly, six placentas had one or more infarctions, of which three had acute atherosis.) Our study demonstrated that increasing the sample size did increase the number of positive cases, as would be expected in a focal lesion. Since three cases had two positive slides out of 4 samples, and 19 cases had no positive slides out of 4 samples, our results suggest a real difference in the density of the lesion among patients. We could not conclusively answer if there were cases that truly had no atherosis. We decided to try a different approach of identifying acute atherosis grossly, which will be described below.
The incidence of decidual vasculopathy varies among investigators, in part because not all studies may include all three potential components, fibrinoid necrosis of the vessels, lipid laden cells in the necrotic material, and separately muscular hypertrophy. I have not discussed the latter because while tempted at times to diagnose the lesion, I have hesitated because quantitative lesions are difficult to judge, because my subjective experience is that the thickness is more in younger gestation placentas, and because I not sure that I am not looking at radial arteries or a transient change in a vessel. However, most studies have concentrated on the two lesions that I am lumping for this web page as acute atherosis. The rational for an overall designation of decidual vasculopathy can be found in the placenta consensus conference monograph15.
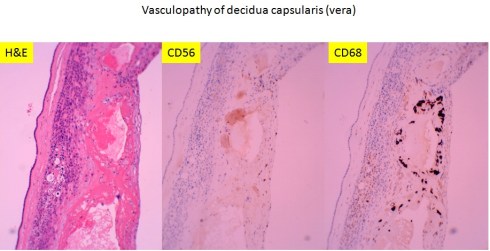
Recently I received an email from Dr. Peilin Zhang with some publications he had written about acute atherosis18(http://dx.doi.org/10.1101/293027.). He demonstrated cytokeratin (AE1/AE3) and , CD56 staining in the foam cells in acute atherosis. He argued that the assumption that the lipid cells in acute atherosis were macrophages was based on morphologic analogy with atheromas in arteriosclerosis and by CD68 staining, and is wrong. I have to admit that CD68 is not specific for macrophages, and in fact I had often seen that is was immuno-positive in cytotrophoblast cells (Fig 34).
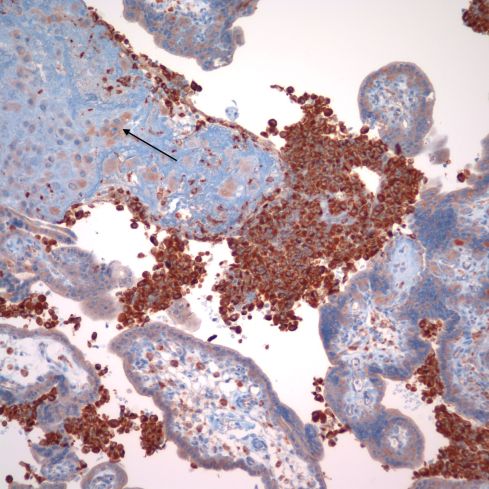
Fig 34: This image of placenta demonstrates massive chronic intervillositis with the many monocytic cells stained deeply dark brown for the antigen CD68. The arrow demonstrates the paler, but still positive for CD68 cytotrophoblastic cells embedded in the surrounding fibrinoid. (CD68, 20x)
I have never tried to identify the cells in acute atherosis with cytokeratins or CD56. However, as I suspected from his publication photograph, and even more clearly in his preprint, the vessels he illustrates are surrounded by cytotrophoblast. I would have considered them partially remodeled vessels in the basal decidua, and not acute atherosis. His prevalence of the acute atherosis/fibrinoid necrosis is high (63%) and from the published illustrations, he evaluated basal decidua.
The consensus monograph of placental diagnosis allows inclusion of unremodeledspiral arteries in the base of the placenta. I only make the diagnosis of acute atherosis in vessels at the base of the placenta if they clearly are not in proximity of cytotrophoblast. In almost all cases that I have diagnosed as acute atherosis, I saw the lesion in the fetal membranes. Given the wide range of reported prevalence of acute atherosis, I suspect that there is poor concordance among pathologists for the diagnosis of acute atherosis in the base of the placenta. It would be interesting to know if the pink hyaline is different in remodeled arteries, which apparently includes fibrin, degenerated trophoblast, and likely secreted cytotrophoblast matrix, from that in acute atherosis 10. A reliable immuno-histological stain to distinguish the two processes would be diagnostically useful.
The importance of the distinction between acute atherosis and trophoblast remodeled vessels is more than academic. This concern is evidenced in a study that compared hemodynamic measurements in preeclamptic mothers seven months after delivery with and without decidual vasculopathy19. The study definition of decidual vasculopathy was “vascular fibrinoid necrosis and lipid filled foam cells in the vascular wall of spiral arteries in either the decidua basalis or parietalis.” The basalis refers to the arteries under the placenta, and parietalis to those in the fetal membranes excluding placenta. This inclusion of both decidual regions likely accounts for the high incidence of 53.4% decidual vasculopathy in their patients and likely includes some specimens with only basilar trophoblastic remodeling of arteries. Importantly, using this definition they found significant differences in some cardiovascular parameters between those patients with and without decidual vasculopathy. The long-term significance of decidual vasculopathy to the mother’s health could not be determined from this study, but the authors demonstrated an increase in hypertension in these patients, and in an earlier publication correlated decidual vasculopathy with some measures of more severe obstetrical disease, including higher diastolic blood pressure, lower birth weight, and lower umbilical cord pH20-22. Their findings may be partially confounded by bundling two distinct histologic entities.
What is the correct definition of decidual vasculopathy? These clinical papers and Dr. Zhang’s paper led me to question the criteria that I use for the diagnosis of acute atherosis, or as Dr. Zhang terms it classic decidual vasculopathy (This excludes muscular hypertrophy as part of the diagnosis). I think a consensus study of shared slides and multiple placental pathologists would better define the nosology of the lesion. I think we may need either some new definitions or a broader classification. I am particularly interested in the relation of lesions seen in the basal placenta to those in the membranes. Are the lesions in the base of the placenta the best indicator of poor spiral artery remodeling? Is acute atherosis seen in the free fetal membranes not a unique lesion, but a failed trophoblast modification of a spiral artery far from the implantation?
The last question was suggested to me in a preliminary, unpublished study of spiral arteries in decidual membrane that I did again with Yasser Daoud. The study attempted to improve our ability to sample acute atherosis in the fetal membranes. The entire membranes after removal from the placental margins were stretched over white cardboard cards, and a dissecting microscope was used to reveal the irregular spiral arteries. We sampled the visible spiral arteries for histological examination and made note of dilated segments that were suspected of being acute atherosis (35).
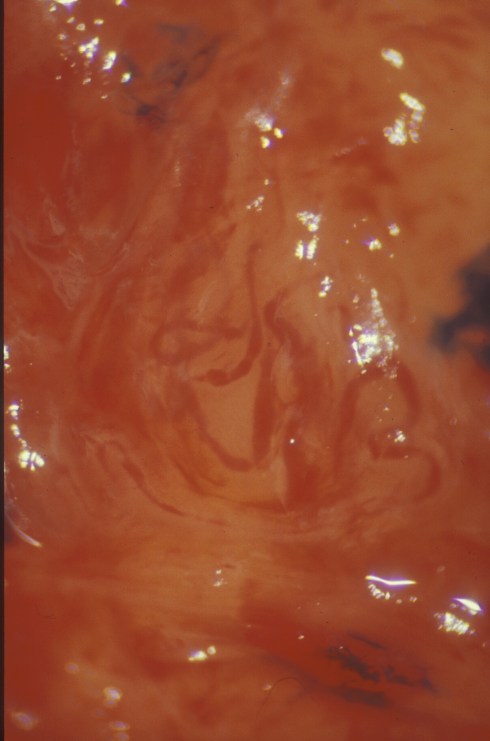
Fig 35: This is a membrane laid flat demonstrating a spiral artery with focal dilatation and hemorrhage.
We found that even in this sample of free fetal membranes, a lesion suspected to be atherosis could be incomplete trophoblast remodeling (Figs 36-37).
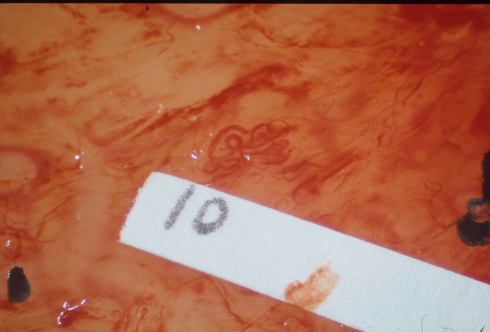
Fig 36: Similar to the sample in slide 35 there is a spiral artery with a dilated segment. The sample was labeled 10 for identification of the corresponding microscopic sample.
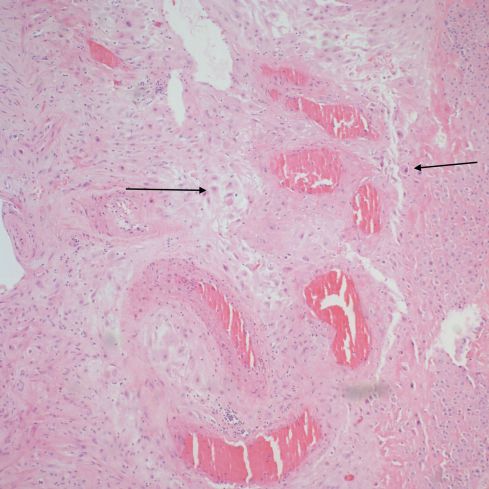
Fig 37: The vessel identified as number 10 demonstrates dilated segments of spiral artery from medial muscle destruction, but this is due to cytotrophoblast that has migrated into the parietal decidua (arrows). (H&E, 10x)
If anyone wants to complete such a study, one measure to record is the distance of the sampled vessel from the placenta. This could be an important parameter determining the prevalence of decidual vasculopathy.
The explanation of the absence of acute atherosis in some patients with toxemia could also be that since acute atherosis is not directly involved in the pathogenesis of toxemia, different patients may have inherent differences in the susceptibility of their spiral arteries to the lesion. Another simple alternative explanation is that those with atherosis may have a different response to the incomplete spiral artery remodeling with perhaps of a different mix of anti-epithelial signals. The pathogenesis of toxemia will be discussed in a later section.
Does acute atherosis have the same pathogenesis in all cases?
We don’t know what causes acute atherosis in toxemia, but the pathogenesis may not be unique to that disease. A different disease might produce the lesion by the same pathologic mechanism or it might through a different pathogenesis produce an appearance that mimics the pathology seen in toxemia. While I was still a fellow in pediatric pathology I examined a placenta from a mother with a clinical diagnosis of TTP that had pink focal lesions of the inner surface of spiral arteries that we suspected were due to small platelet clots on the endothelium causing necrosis of the adjacent media. We sent the slides to a respected consultant who said we were just seeing acute atherosis. I still have my doubts but I cannot find images from that case. Others have since reported TTP as an acute atherosis mimic23. In other cases, I have seen necrotic vascular lesions that are of unknown etiology even in normal pregnancies that I have diagnosed as fibrinoid necrosis of spiral arteries for lack of a better term. These lesions could have a different etiology from those in toxemia. However the resemblance still needs an explanation (Figs 38,39).
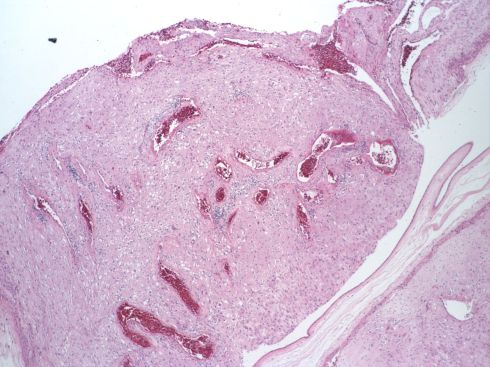
Fig 38: This low power image of the membranous decidua sectioned tangential to the membrane plane shows in the convoluted cross sections of a spiral artery, which appears dilated in the right corner of the field. The placenta was uncomplicated with a 3000 kg infant at term with no maternal history of toxemia or autoimmune disease. (H&E, 4X)
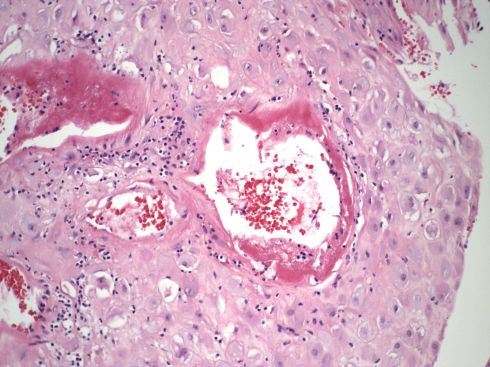
Fig 39: This is a high power of the above section showing what appears to be either fibrin or fibrinous necrosis but certainly thinning and dilatation of the artery wall. (H&E 20x)
In acute atherosis with fetal growth restriction, the question I think becomes, at least in infants whose placenta shows evidence of utero-placental ischemia, why they do not have clinical toxemia. In diabetic mothers with acute atherosis a mimic lesion from endothelial injury from glucose arises as a possibility. In immune complex disease such as SLE the mechanism is again likely to be endothelial injury with an unexplained predilection fro spiral arteries. Studying in detail the differences in acute atherosis with different diseases potentially could yield new insights into the pathogenesis of the lesion.
Do the natural killer cells that are a normal component of endometrium have any involvement in the formation of acute atherosis?
In the papers that Dr. Zhang sent to me, he argues that CD56 is key to acute atherosis, and that the antigen is in some way transferred from CD56 positive natural killer cells. There certainly is a literature on the role of natural killer cells in pregnancy, and Dr. Zhang reviews some interesting studies in mice. I recall a paper presented at a symposium at the University of Kentucky on comparative studies of placenta on the importance of natural killer cells in pig pregnancies. In the Guinea pig, the mesometrial arteries are remodeled by non-trophoblastic mononuclear cells 24. In vitro research points to matrix degrading proteases produced by natural killer cells that migrate to spiral arteries in the decidua 25. However, I currently cannot accept the CD56 vasculopathy hypothesis, but it made me consider two things. The first is that anti-CD56 marks cytotrophoblast is a not a surprising finding, given that this is a common surface antigen of embryonic tissue and has been known since at least 1994 26. However, it may be important that CD56 is a cell adhesion molecule (once thought to neuro-specific, hence the alternative name, N-CAM), and that it can bind to itself. The first consideration is can natural killer cells bind to cytotrophoblast via CD56?
The second consideration is the potential value of understanding the evolutionary play between invasive trophoblast cells, which mimic malignancy in many ways, and the role of natural killer cells. I agreed with Kurt Benirschke that we needed to better understand the evolutionary biology of the placenta from pre-placenta forms for example guppies to the wide variation in placentation in mammals. He was way ahead of me, and years before his recent death, he started an important web site of comparative placentation. The evolution of natural killer cells in the endometrium might shed light on their role in human pregnancy and malignancy.
Do toxemia patients with acute atherosis have more severe obstetrical complications or different time of onset of disease?
Toxemia is often separated into an early onset versus a late onset usually milder disease. The major complications in the mother are eclampsia (seizures), liver and cerebral hemorrhage, and placental abruption. The fetal complications are growth restriction and its potential complications including stillbirth. Some studies have found an increased incidence of complications with acute atherosis.
What happens to the trophoblast remodeled arteries that are not shed with the decidua?
Pathologists see the residua of previously remodeled spiral and radial arteries in uteri removed for other causes later in a gravid woman’s life. (Spiral arteries in the superficial decidua are shed at the end of delivery.) How is this process of involution controlled? Is it simply the removal of progesterone or other signals that were associated with the placenta? How are new radial and deep spiral arteries regenerated? As per Dr. Zhang, is there a role of natural killer cells in removing intravascular cytotrophoblast, since both they and the cytotrophoblast share an adherence molecule, CD56?
These questions bear on the question as to why primigravidas are at the highest risk of toxemia. As a resident, I recall speculation that it must be due to acquired immuno-tolerance. Some papers even suggested that the risk increased with a different father of the fetus. However, that notion was dampened by a Norwegian epidemiologic study published in the New England Journal in 200227which demonstrated the interval between pregnancies was the determining factor. A related phenomenon is that the firstborn child usually has the lower birth weight of his/her siblings. To most observers, this suggests that even without diagnostic toxemia, the firstborn child likely is less successful establishing a utero-placental circulation than his later siblings. Dr. Yee Khong who made important contributions to the concept of inadequate remodeling of spiral arteries to the development of toxemia, tried to understand this primigravida phenomenon by a direct observation. He and colleagues using a random sample from the superficial myometrium of non-gravid hysterectomy specimens, compared the extent of smooth loss in the media, and of elastic tissue fragmentation of spiral arteries in women of different gravida28. They found significant evidence of elastica degeneration and loss of medial muscle between nulliparous women and those who had children. As noted in the paper, they could not determine if the sampled arteries had been beneath the placenta. The assumption was that within sampling error, the effect was still evident because the arteries had been modified in the past by trophoblast remodeling within the placental bed. If prior pregnancy weakens spiral arteries and accounts for larger fetal growth and less toxemia in subsequent pregnancies, is it possible that the extent of deep trophoblast remodeling of spiral arteries extends beyond the immediate placental bed. It would be interesting to plot not only the extent of finding cytotrophoblast beyond the placental margin, but also the pattern of vascular remodeling in uteri removed after pregnancy for disease unrelated to pregnancy.
Bottom line:
The spiral artery remodeling is a very complex process but critical to lowering vascular resistance and allowing the necessary increase in uterine blood flow to the placenta. Toxemia is likely due to a primary failure to obtain sufficient high flow through the intervillous space. The basis for this failure is still moot, but the process may be initiated early in pregnancy. One common consequence is the placental and fetal responses to decreased utero-placental blood flow, which will be discussed in Section 9c and 9E. The placenta in toxemia induces active signals in the maternal blood that oppose endothelial growth factor and results in damage endothelial cells. The lesions in the mother will be discussed in Section 9b.
There are many unanswered questions relating the spiral artery lesions to toxemia. Why is toxemia more common in the first pregnancy and in molar and diandric partial (triploid) molar pregnancies? What is the relationship of acute atherosis to spiral artery remodeling and toxemia? How does the failure of trophoblast remodeling of spiral arteries predispose to placental infarction and retroplacental hematoma?
For the diagnostic pathologist, the major question is whether any lesion, acute atherosis or otherwise, has any prognostic value for future pregnancies or future cardiovascular disease in the mother. Prospective long-term clinical pathological correlation studies are needed to answer this question. These studies need to utilize accepted pathological criteria. Further basic molecular and experimental studies of trophoblastic remodeling of vessels is likely to provide valuable insights not only into toxemia, and utero-placental ischemia, but also into cellular pathways of trophoblastic invasion that could be coopted in malignancy.
References:
- Ramsey EM. The vascular pattern of the endometrium of the pregnant rhesus monkey (Macaca mulatta). Carnegie Intitute Washington Publication 518 1949;33:113-48.
- Ramsey EM, Donner MW. Placental vasculature and circulation. Philadelphia: W. B. Saunders Company Ltd; 1980.
- Degner K, Magness RR, Shah DM. Establishment of the Human Uteroplacental Circulation: A Historical Perspective. Reprod Sci 2017;24:753-61.
- Panigel M. The human placenta. Anatomy and morphology. Clin Obstet Gynaecol 1986;13:421-45.
- Boyd JD, Hamilton WJ. The Human Placenta. Cambridge: W. Heffer & Sons LTD; 1970:209.
- Pijnenborg R, Vercruysse L, Hanssens M. The uterine spiral arteries in human pregnancy: facts and controversies. Placenta 2006;27:939-58.
- Bendon RW, Mimouni F, Khoury J, Miodovnik M. Histopathology of spontaneous abortion in diabetic pregnancies. Am J Perinatol 1990;7:207-10.
- Brosens I, Robertson WB, Dixon HG. The physiological response of the vessels of the placental bed to normal pregnancy. J Pathol Bacteriol 1967;93:569-79.
- Sheehan HL, Lynch JB. Pathology of Toxaemia of Pregnancy. Baltimore: The William and Wilkins Company; 1973.
- De Wolf F, De Wolf-Peeters C, Brosens I. Ultrastructure of the spiral arteries in the human placental bed at the end of normal pregnancy. Am J Obstet Gynecol 1973;117:833-48.
- Whitley GS, Cartwright JE. Cellular and molecular regulation of spiral artery remodelling: lessons from the cardiovascular field. Placenta 2010;31:465-74.
- Aberdeen GW, Bonagura TW, Harman CR, Pepe GJ, Albrecht ED. Suppression of trophoblast uterine spiral artery remodeling by estrogen during baboon pregnancy: impact on uterine and fetal blood flow dynamics. Am J Physiol Heart Circ Physiol 2012;302:H1936-44.
- Cole LA. Hyperglycosylated hCG. Placenta 2007;28:977-86.
- Stanek J. Histological Features of Shallow Placental Implantation Unify Early-Onset and Late-Onset Preeclampsia. Pediatr Dev Pathol 2019;22:112-22.
- Morgan TK PW. Pregnancy-Induced Uterine Vascular Remodeling and the Pathophysiology of Decidual Vasculopathy
. In: Khong TY ME, Nikkels PGJ, Morgan TK, Gorgdijn SJ, ed. Pathology of the Placenta: A Practical Guide. Switzerland: Springer; 2019:221-31.
- Labarrere CA. Acute atherosis. A histopathological hallmark of immune aggression? Placenta 1988;9:95-108.
- Kitzmiller JL, Watt N, Driscoll SG. Decidual arteriopathy in hypertension and diabetes in pregnancy: immunofluorescent studies. Am J Obstet Gynecol 1981;141:773-9.
- Zhang P. Decidual Vasculopathy in Preeclampsia and Spiral Artery Remodeling Revisited: Shallow Invasion versus Failure of Involution. AJP Rep 2018;8:e241-e6.
- Stevens DU, Al-Nasiry S, Fajta MM, et al. Cardiovascular and thrombogenic risk of decidual vasculopathy in preeclampsia. Am J Obstet Gynecol 2014;210:545 e1-6.
- Stevens DU, Al-Nasiry S, Bulten J, Spaanderman ME. Decidual vasculopathy in preeclampsia: lesion characteristics relate to disease severity and perinatal outcome. Placenta 2013;34:805-9.
- Stevens DU, Al-Nasiry S, Bulten J, Spaanderman ME. Decidual vasculopathy and adverse perinatal outcome in preeclamptic pregnancy. Placenta 2012;33:630-3.
- Stevens DU, Smits MP, Bulten J, Spaanderman ME, van Vugt JM, Al-Nasiry S. Prevalence of hypertensive disorders in women after preeclamptic pregnancy associated with decidual vasculopathy. Hypertens Pregnancy 2015;34:332-41.
- Jamshed S, Kouides P, Sham R, Cramer S. Pathology of thrombotic thrombocytopenic purpura in the placenta, with emphasis on the snowman sign. Pediatr Dev Pathol 2007;10:455-62.
- Hees H, Moll W, Wrobel KH, Hees I. Pregnancy-induced structural changes and trophoblastic invasion in the segmental mesometrial arteries of the guinea pig (Cavia porcellus L.). Placenta 1987;8:609-26.
- Harris LK. IFPA Gabor Than Award lecture: Transformation of the spiral arteries in human pregnancy: key events in the remodelling timeline. Placenta 2011;32 Suppl 2:S154-8.
- Burrows TD, King A, Loke YW. Expression of adhesion molecules by endovascular trophoblast and decidual endothelial cells: implications for vascular invasion during implantation. Placenta 1994;15:21-33.
- Skjaerven R, Wilcox AJ, Lie RT. The interval between pregnancies and the risk of preeclampsia. N Engl J Med 2002;346:33-8.
- Khong TY, Adema ED, Erwich JJ. On an anatomical basis for the increase in birth weight in second and subsequent born children. Placenta 2003;24:348-53.
Comment
Dr. Peilin Zhang has contributed the following comments on this page.