Fibrinoid
The pathologist looking at any placenta finds eosinophilic amorphous material in the intervillous space. Like a cave, some clings to the underside of the fetal surface with stalactite extensions toward the maternal surface, or mounds on the maternal floor like stalagmites or even forms columns from top to bottom (fig 1).

Fig 1
Other less cellular accumulations form shapes that mimic the pattern of villi (fig 2).

Fig 2
The amount of such eosinophilic material varies from placenta to placenta with no salient clinical correlation.
What is this eosinophilic amorphous material that is often called fibrinoid? Dr. Peter Kaufmann and associates, as well as others, have identified two separate forms using immunohistologic staining and electron microscopy1. The first is a dense acellular type that they have designated as fibrinous fibrinoid. It contains fibrin, platelet fragments and likely fragments of degenerated syncytiotrophoblast2, 3. It is usually found on the surfaces facing the intervillous blood flow. This is not notably different from a mural thrombus in an artery and it seems reasonable to refer to this simply as fibrin. If the fibrin is on the surface a villus, then it is designated perivillous fibrin (fig 3).

Fig 3
The second type of fibrinoid is a looser, less homogenous substance with cytotrophoblast embedded within it (fig 4).
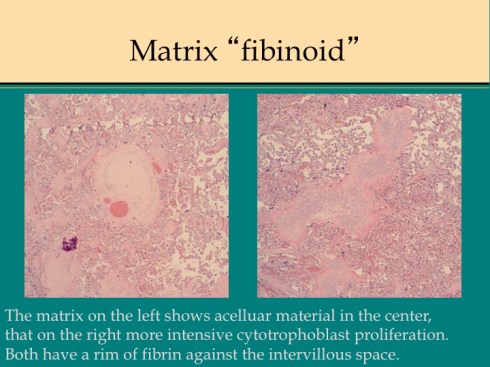
Fig 4
Chemically the material is a type of extracellular matrix produced by the cytotrophoblast4-9. There may be a layer of fibrin between this matrix and the intervillous blood flow, but fibrin is not integral to this type of fibrinoid. This can be called matrix type fibrinoid, but is simply cytotrophoblast proliferation with matrix secretion. The pathologist usually sees both types of fibrinoid together in intimate spatial relationships that beg for further explanation.
Fibrin deposition in the placenta is derived from blood. Blood clotting is the precipitation of insoluble, sticky fibrin from the cleavage of soluble fibrinogen by the enzyme thrombin. Numerous interacting factors including platelet degranulation regulate thrombin activity. Fibrin deposition is counteracted by the cleavage of fibrin by plasmin. In turn plasmin production is regulated by multiple factors. The basic biology of blood clotting is well understood in blood vessels in which patency is maintained by the endothelium. More than a century ago Virchow noted that fibrin deposition in blood vessels, that is thrombus formation, occurs in response to blood stasis, tissue injury and/or thrombophilia. The latter can be caused by stress notoriously after injury such as surgical procedures or be an innate condition such as genetic mutation. The reader can find the details in textbooks of hematology or pathology.
The intervillous space is in effect a blood vessel but instead of blood flowing into a smooth cylindrical channel it flows through a mass of projecting root-like villi and instead of having a lining of endothelium, it is lined by syncytiotrophoblast acting as the endothelium (among its many functions). Unlike the streamlined blood flow in most of the vascular system, the intervillous blood flow is a fountain of blood sprayed into a space filled with villous baffles. The monograph by Elizabeth Ramsey, Placental Vasculature and Circulation, gives a clear and cogent presentation of this flow10. This inherently turbulent and irregular blood flow provides an explanation for the normal presence of fibrin in the placenta. As in the vegetative fibrin deposition that forms on damaged heart valves (marantic endocarditis), turbulence creates eddies with areas of stasis and hypoxic injury favor fibrin formation. Some authors have proposed the interesting, but difficult to prove, hypothesis that the filling in by fibrin of this intervillous dead space helps streamline flow and improve perfusion efficiency11. Under the microscope, perivillous fibrin can look remarkably in pattern like extensions and bridges between villi that mimic the normal villous architecture. As occurs in vitro, the fibrin is often resurfaced with syncytiotrophoblast completing the disguise3, 12(fig 3). Could this pseudovillous pattern reflect conditions of stable flood flow in the intervillous space?
The initiating event in perivillous fibrin deposition is likely syncytiotrophoblast injury. Syncytium adjacent to fibrin deposition demonstrates morphological and biochemical evidence of injury usually with apoptosis, programmed cell death13-16. There is evidence that hypoxia can initiate apoptosis in syncytiotrophoblast17-19. In routine pathology, syncytiotrophoblast nuclei near or within the formation of perivillous fibrin demonstrate hyperchromatic irregular nuclei typical of apoptosis (fig 5).

Fig 5
If injury decreases antithrombogenic activity or exposes the basement membrane that underlies the synctiotrophoblast to the maternal blood, thrombin will be generated, and fibrin deposited.
A placental infarction provides a model of hypoxic placental injury that elucidates fibrin deposition (fig 6).

Fig 6
A true placental infarction occurs when the flow from a spiral artery is occluded and the overlying villi and intervillous space cease to have blood flow. The center of the infarction demonstrates collapse of the intervillous space and coagulation necrosis of villi, the morphologic features of anoxic cell death. An infarction becomes surrounded at its margin with villi trapped in perivillous fibrin. At the far periphery, villi demonstrate an increase in syncytial knots (piled up nuclei) and capillary syncytial membranes (thinned syncytium above fetal capillaries), both part of the adaptation of villi to ischemia that increases oxygen diffusion from mother to fetus. The reasonable assumption is that an infarction creates a gradient of hypoxia most severe at the center of the infarction. This assumption provides a simple paradigm for interpreting the morphologic response of the placenta to hypoxia. The most severe hypoxia/anoxia results in coagulation necrosis, an intermediate degree results in syncytial necrosis and pervillous fibrin deposition often with survival of Langhan’s cytotrophoblast that normally underlie and regenerate the syncytium, and the least hypoxic injury results in syncytial knot formation. This paradigm explains the deposition of fibrin in areas of poor perfusion around the periphery of the normal fountain of blood entering from the spiral artery as occurs beneath the fetal surface of the placenta. This does not imply that hypoxia is the only mechanism that induces syncytial injury and apoptosis.
Authors have theorized that fibrin can deposit on intact syncytiotrophoblast and cause hypoxic injury20. The syncytiotrophoblast normally has strong plasminogen activity that upregulates in the presence of fibrin21. A potent thrombogenic stimulus could still overcome this activity and layer fibrin on intact syncytiotrophoblast. The appearance of such deposition is seen with laminated intervillous thrombi. However, perivillous fibrin deposition is not increased in mothers with inherited thrombophilia. Placentas from these affected mothers may have infarctions from spiral artery thrombi, but they do not have massively increased perivillous fibrinoid. I have not found convincing studies that show a benefit of maternal anticoagulation in preventing recurrence of massively increased perivillous fibrinoid 22, 23. Subtle syncytial injury, for example a factor causing decreased plasminogen receptor, could lead to fibrin deposition that in turn causes hypoxia and necrosis of the syncytium resulting in the typical appearance of perivillous fibrinoid deposition on denuded basement membrane. The morphologist might have trouble telling the difference especially if apoptosis was being induced in surrounding nuclei.
The proliferation of cytotrophoblast and deposition of matrix may be a secondary process if syncytiotrophoblast dies, but cytotrophoblast survives. Released from syncytiotrophoblast relationships, the cells may revert to a different phenotype and lay down matrix much as they do at the interfaces of primary cytotrophblast with maternal tissue. In vitro, cytotrophoblast remain single cells at 1% oxygen, but form syncytium at 20%24. The matrix is the glue that holds the placenta to the decidual stroma25. Other cytotrophoblast cells in the intervillous space are likely residual from the original columns between lacuna of the developing placenta. Matrix in the placenta is always associated with cytotrophoblastic cells, but very often has a surface of fibrin. There are also intermediate forms in which villi encased in fibrin show a subtle proliferation of cytotrophoblast around there periphery (fig 7).

Fig 7
Cytotrophoblast proliferation and matrix secretion could be interpreted as the default program for cytotrophoblast freed from the villous architecture.
There are many unanswered questions about normal fibrinoid deposition. How can a focal area of syncytiotrophblast, which is a continuous cell sheet, be injured without broad damage? Why does perivillous fibrin take the geometrical form that it does? Why is more fibrin not deposited in placentas of mothers with thrombophilia? Is there such a thing as intravillous fibrinoid or this an illusion of syncytiotrophoblast “re-endothelializing” the surface of fibrin? The small punctuate intravillous fibrinoid described by Fox in cases of potential immune complex disease with both Rh sensitization and with anti-insulin antibodies could be a different and today uncommon entity26. Is there cross talk between the syncytiotrophoblast and the fetal circulation? For example, stem villi often have perivillous fibrin instead of syncytiotrophoblast on the surface. This appears to be beneficial since high energy syncytiotrophoblast transport would not find fetal capillaries on the villous side to carry away the transported chemicals. Perhaps accumulation of these transported products leads to syncytiotrophblast degeneration. On the other hand, villi trapped in fibrin (or in an infarction) loose their fetal circulation, as if some transport product from syncytiotrophblast is needed to maintain the fetal circulation.
Bottom line: For practical descriptive purposes, the term “fibrinoid” in the placenta meets the need to describe focal accumulations of fibrin, cytotrophoblast proliferation and matrix deposition. Fibrinoid is understood to be composed of varying proportions of fibrin and matrix components. The most likely relationship between the components is that syncytiotrophoblast injury promotes fibrin deposition on the exposed villous surface. The underlying cytotrophoblast then proliferates and may produce matrix. In other cases, non-villous cytotrophoblast may proliferate and the exposed matrix will initiate fibrin deposition. In certain contexts, such as interpreting the pathophysiology, more precision may be had by referring directly to fibrin deposition or matrix proliferation.
Focal massive pervillous fibrinoid deposition
A curious phenomenon is the formation of a focal area of massive perivillous fibrinoid. This lesion has a superficial gross resemblance to placental infarction demonstrating a solid white to yellow mass lesion. The lesions can be grossly distinguished in that close inspection of an infarction shows villi in a collapsed intervillous space, but fibrinoid deposition shows a solid rubbery mass often with sponge like channels of blood flow (fig 8).

Fig 8
The massive fibrinoid deposition strangles encased villi that then undergo the same process of coagulation necrosis and loss of fetal circulation as in an infarction. Fox attempted to find any clinical correlation for these focal lesions, and did not find one27.
Such a focal lesion is unlikely to be due to a circulating factor in maternal blood. Focal placental mosaicism can not be totally excluded, but has never been demonstrated. The sharply demarcated, varyingly sized, contiguous area often involves an extent supplied by multiple spiral arteries. Some small, often marginal, lesions show a small infarction buried within the fibrinoid shell. In such cases, the lesion appears to be a very exaggerated periphery of an old infarction. The relationship of focal massive perivillous fibrin deposition to diffuse massive perivillous fibrin remains unknown.
MFI: Massive perivillous fibrinoid infiltration/ maternal floor infarction of the placenta
This lesion or lesions has proved difficult to define. Infarction normally refers to occlusion of an end artery to a portion of tissue with subsequent ischemic necrosis (death) of that tissue, most notably occurring in the brain or heart, but also in the placenta. Maternal floor infarction then logically should refer to obstruction of blood flow of the arterial entrance to the intervillous space causing necrosis of the overlying placenta. Thickened fibrin or extravillous matrix at the interface of placenta and basal decidua could produce such an infarction. Microscopically, an infarction of the placenta would show collapse of the intervillous space with coagulation necrosis of the villi. A maternal floor infarction then should show fibrin or matrix thickening of the maternal surface with otherwise typical changes of infarction in the villi. It is moot whether such a lesion which I will call true maternal floor infarction occurs, but the term maternal floor infarction is not usually used in this sense. Instead it refers to extensive gross thickening of the maternal surface of the placenta with fibrin or matrix, often in the context of fetal growth retardation or death. Focal thickening of the maternal floor is common and not usually considered a pathological lesion. Diffuse deposition of fibrin/ matrix may be isolated to the maternal floor, but more commonly is associated with increased perivillous fibrin/matrix deposition throughout the placenta.
Maternal floor infarction was perhaps first introduced in the English published literature in the textbook by Benirschke and Driscoll who presented a case of a woman with nine recurrences of the lesion, each resulting in stillbirth. In a subsequent paper of 60 cases with Dr. Benirschke as an author, the definition of MFI was given as “ marked increase in fibrin deposition involving the decidual floor with villous encasement”28. Others have tried to define the same lesion. Another study identified 25 patients as having maternal floor infarction based on “massive perivillous fibrin deposition proposed by Fox,” and elaborating “marked fibrin deposits among the villi when these deposits obliterated over three fourths of the intervillous space in all three sections”22. Another review of 80 placentas with MFI used the following definitions “ classic MFI (basal villi of the entire maternal floor encased by perivillous fibrinoid of > 3 mm thickness on at least one slide); transmural MFD (perivillous fibrinoid material extending from the maternal surface to the fetal surface, encasing >50% of the villi on at least one slide); borderline MFD (25%-50% villi on at least one slide encased by perivillous fibrinoid material in a transmural or nearly transmural, distribution)”29. Of note, a single recurrence occurred in a mother who initially presented with transmural MFD, but recurred with maternal floor infarction. No matter how precise or vague the definitions, studies of the lesion have demonstrated correlations of MFI with intrauterine growth retardation, fetal death, and recurrence in subsequent pregnancies. The lesion may encompass functionally or biologically related entities, like an enclosure with zebras, horses, and camels, which will eventually be better defined by pathogenesis.
A diffuse increase in perivillous fibrinoid is a marker of significant pathology. In my files, I code the lesion MFI, an acronym that includes massive fibrinoid infiltration or maternal floor infarction. This preserves the acronym, but “infiltration” seems less accurate than “deposition”. The lesion presents on gross examination as a tactile resistance to compression of the placenta which is normally soft and flabby. The maternal surface often has a firm white convoluted pattern which on cut section may be continuous with diffuse increased fibrinoid or appear accentuated along the base (fig 9).

Fig 9a

Fig 9b
The pattern of parenchymal fibrin deposition varies. Some placentas have a diffusely granular network of fibrinoid placenta (fig 10).

Fig 10a
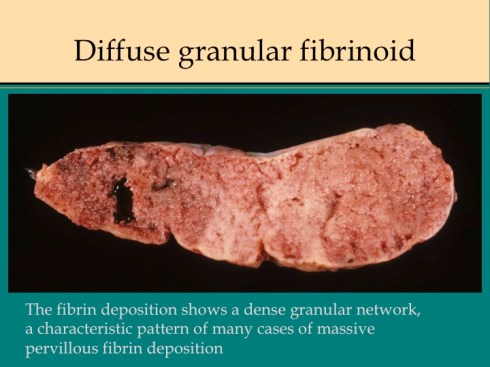
Fig 10b
Channels of open blood flow often appear embedded in the more confluent fibrinoid deposition. These channels may demonstrate laminated thrombi likely the result of slow flow and turbulence analogous to venous thrombi. Other placentas have a lumpy, coarsely irregular pattern that sometimes exaggerates the stalactite pattern that can be imagined to surround the jets of arterial inflow (fig 11).

Fig 11a

Fig 11b

Fig 11c
There are rare cases that have increased fibrinoid confined to the placental floor with overlying ischemic infarction which could be considered true maternal floor infarction (fig 12).

Fig 12a

Fig 12b
These cases may have a different etiology from those with increased perivillous fibrinoid. Increased perivillous fibrinoid deposition is a continuum and likely, not all pathologists would agree on which placentas qualify for the diagnosis of MFI.
Under the microscope, massive increased perivillous fibrinoid in some cases shows a collapsed intervillous space and perivillous fibrin deposition showing the same features as in uncomplicated pregnancies, with apoptosis of syncytiotrophoblast, denuded villi with viable cytotrophoblast and matrix, and fibrin enmeshed sclerotic/ necrotic villi. The villi may appear necrotic or may have persistent or even somewhat proliferative cytotrophoblast beneath the fibrin (fig 13).
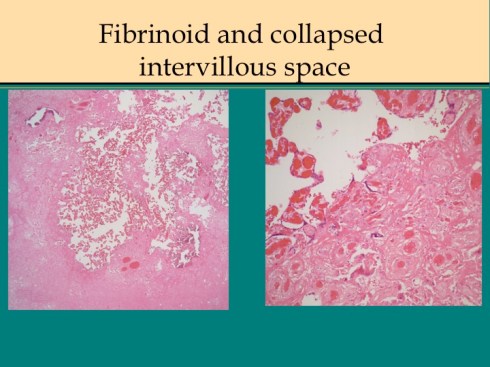
Fig 13a

Fig 13b
This pattern is often seen with the less confluent fibrin deposition. The same pattern can be seen in presumed ischemic areas of intervillous collapse and villous agglutination in placentas that are othwise normal from normal pregnancies. In a few placentas, often with the granular network of fibrin, there is a prominent proliferation of cytotrophobast surrounded by fibrin (or focally even without a fibrin surface)(fig 14).
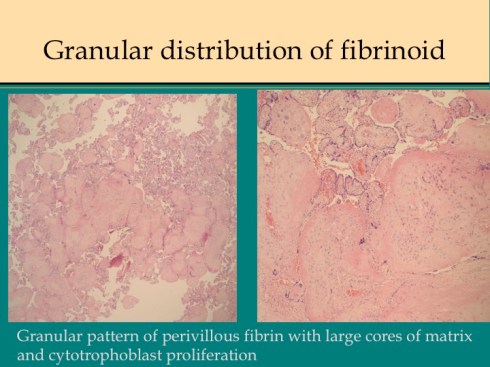
Fig 14a

Fig 14b
This is the same appearance that may be seen on the floor of the placenta or in cytotrophoblastic “X-cell” collections in normal placentas.
The two patterns of coarse versus granular fibrinoid deposition could present ends of a spectrum of two different pathogenetic mechanism. The coarse deposition would result from processes such as stasis or turbulence that favors collapse of the intervillous space and fibrin deposition. The mechanism is analogous to the fibrinoid deposition around the periphery of a placental infarction. The granular distribution would be typical of a serum factor that would diffusely injure syncytiotrophoblast resulting not only in a more even distribution but in more cytotrophoblast proliferation following injury restricted to overlying syncytium. The cytotrophoblast normally replenishes the syncytium. The syncytium is extraordinarily metabolically active with high oxygen and glucose consumption which could make it vulnerable to toxic or metabolic injury. This mechanism was demonstrated experimentally by administering monoiodineacetate, an inhibitor of carbohydrate metabolism to pregnant guinea pigs which produced a lesion of syncytial destruction and fibrinoid (bundles of fibers on EM not identified immunologically) filling the labyrinth of the placenta in a description that could pass for MFI30. A similar mechanism could underlie cases of MFI reported with long chain hydroxyacyl-coenzymes A dehydrogenase deficiency31, 32. The one case of recurrence of MFI that I examined had the diffuse granular pattern in both placentas. One of the cases associated with autoimmune disease also had this pattern. Fibrinoid patterns falling in between would have different degrees of syncytial injury which would also be sensitive to regional blood flow. There is no proof that the dichotomy of fibrinoid patterns reflects different mechanisms, but this notion
Placentas with MFI show varying degrees of villitis and intervillositis. Villitis is common in placentas, but in some placentas areas with villitis correspond to those with perivillous fibrinoid. Even more striking and perhaps more common are placentas in which intervillositis appears to be directly part of the deposition of perivillous fibrinoid (Fig 21).

Fig 21a

Fig 21b

Fig 21c
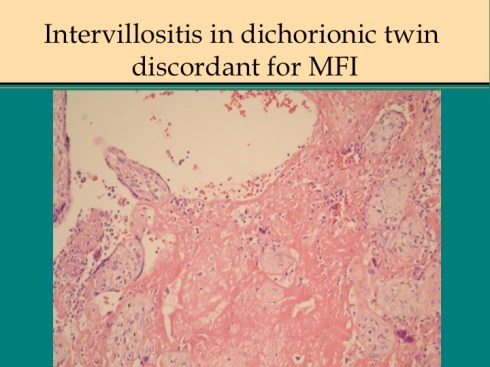
Fig 212d
Whether the presence of the inflammatory cells, usually monocytes, but some neutrophils, is a cause of syncytiotrophoblast injury or a consequence of an immune/thrombogenic response can not be determined from the histology.
Over several years, I had received three placentas with the prenatal ultrasound diagnosis of complete replacement of the placenta by chorangioma that pathologically were consistent with my criteria for maternal floor infarction at that time, namely gross and microscopically increased perivillous fibrin that was not restricted to a single sector of the placenta. I suggested to our university maternal fetal medicine group that they review prenatal ultrasound examinations in placentas with the diagnosis of maternal floor infarction. They did and they agreed that there was a distinctive hyperechogenic ultrasonogrpahic appearance of the placenta in the three index infants, but not in 6 other cases diagnosed as maternal floor infarction. Looking prospectively for these ultrasound features, they correctly diagnosed maternal floor infarction in two pregnancies, one a recurrence. We published this case series 33. Comparing the gross cut sections of the placentas in this study with and without an abnormal ultrasound pattern (Fig 15)

Fig 15a

Fig 15b
, equally varied patterns of fibrin deposition were present in both groups, so it was not the pathological pattern that determined ultrasound detection of the lesion. As a pathologist, I am surprised that the gross alterations of placental consistency and of intervillous blood flow are not always detectable by antenatal ultrasound. The only other published experience found 8 abnormal placentas with antenatal ultrasound from 12 cases with pathologically diagnosed MFI34.
One ultrasonographic feature related to fibrin deposition is the presence of multiple subchorionic cysts (Fig 16).
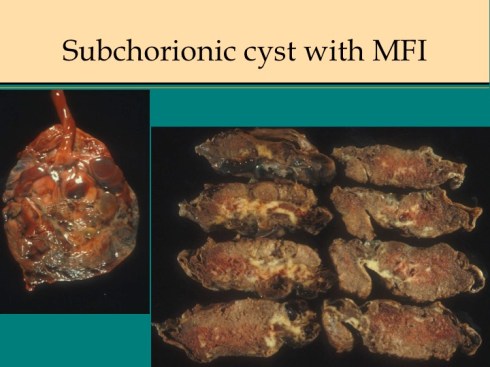
Fig 16
Since subchorionic cysts appear to be due to secretion by cytotrophoblast trapped in subchorionic fibrin beneath the fetal surface, the association may not be accidental. A placental ultrasound correlation study found cysts in 3 of 34 placentas with maternal floor infarction35. The cyst fluid has been demonstrated immunohistologically to contain major basic protein, the same as the toxic component in eosinophil granules36, 37. Perhaps under certain conditions, cytotrophoblast differentiation morphs to that of trophoblast invasion of the maternal tissue with secretion of cytotoxic chemicals. Authors have suggested that this toxic fluid component could play a role in the pathology of MFI, but that role was not clarified38. Another possible prenatal diagnostic clue favoring MFI is loss of bone density in the fetus. Most of my cases with body radiographs do not show obvious hypodensity of bone, but one had rachitic changes.
MFI has been associated retrospectively with varying incidences of fetal growth retardation and fetal death. The pathology suggests that MFI causes loss of function in enmeshed villi, maternal to fetal perfusion mismatch, and sometimes thrombosis of some intervillous space. The perfusion mismatch would occur because more blood from spiral artery flow is shunted to the remaining open intervillous space which has relatively few villi. Logically stillborn infants with MFI should be small for gestation unless the onset of MFI was very rapid, or the death was unrelated to the extant of loss of functional placenta.
Published studies have varying rates of stillbirths associated with MFI placentas from 17% (33/190) (using the definition: “diffuse, firm, pale thickening of the decidual basalis due to the heavy deposition of fibrin in the decidua basalis”)39. to 40% (24/60)28. The incidence of intrauterine growth retardation in MFI patients also varies from 31% (11/56)29 to 54% (19 of 35 livebirths) 28 to 92% (11 of 12 live and stillbirths)33 to 100% (12/12). The association of MFI with growth retardation and fetal death appears significant, but in the larger picture it is an infrequent cause of either. The incidence varies from 0.5% (190/39,215) in the Perinatal Collaborative Study39 with the most inclusive definition, to 0.03% in a different study (13/47,022)40. The incidence of MFI and its complications in retrospective studies could have a selection bias in that patients with severe growth retardation or fetal death are more likely to be seen in tertiary centers and more likely to have their placentas sent to pathology. MFI is less likely to be diagnosed in uncomplicated pregnancies. In our study in which institutions send placentas from all deliveries for examination (see P.R.O.O.F study on web site) only 2 cases of MFI were diagnosed from 6,084 patients (0.03%). One was not small for gestation (assuming gestation was correct) and the other at 27 weeks gestation had no birth weight on the placental pathology history (patient not contacted for consent to the study). There is no quantitative study to answer how much fibrin or what pattern of fibrin deposition is needed to produce fetal compromise. Incidence of complications would also be affected by the definition of MFI used in the study. These same problems would also impact estimates of the incidence of MFI.
The increased risk of recurrence rate in subsequent pregnancies is clinically important. For the individual patient, this can be 100% (as in the nine recurrences in Benirschke and Driscoll). This striking reoccurrence makes a fetal genetic trait unlikely and is more consistent with an allo-immunization (similar to Rh antigen sensitization) or to a persistent maternal disease. The Collaborative Perinatal Study found that 50% of mothers with MFI had prior abortions or stillbirths compared to 27% of controls (P<.001). Most other series of cases report one or two reoccurrences, but the numbers of women followed in subsequent pregnancies is too low for statistical analysis. Given the very low incidence of MFI, it is astronomically unlikely that most recurrence was due to chance. Stillbirth from MFI may be prevented by careful observation of the fetus and placenta in subsequent pregnancies of affected mothers33, 41.
Recurrence points to an underlying fetal or maternal condition causing MFI, but does not point to a specific etiology. Infrequently MFI is associated with other clinical disease that plausibly could cause the lesion. We reported mothers with autoimmune disease and MFI42. Both placentas had confluent deposition (one predominantly fibrin, the other matrix) and were fully developed before midgestation (Fig 17).

Fig 17
This pattern would be consistent with an antibody present from early gestation that could injure syncytiotrophoblast or interfere with surface anti-thrombotic proteins such as plasminogen activator. There are other reports of MFI associated with autoimmune disease. Maternal polymyositis has been associated with MFI 43-46. A study of 22 MFI placentas reported 2 mothers with SLE29. Conversely, a study of 42 patients with SLE reported two cases with perivillous fibrinoid change with growth retardation (the histological figure in the paper is not convincing for MFI). Both of these patients also had lupus anticoagulant/anticardiolipin antibodies. Three patients with anti-phospholipid syndrome were reported to have massive perivillous fibrin deposition47. These cases could be due to an antibody other than the thrombophilic anti-phospholipid, in patients prone to develop autoantibodies, as in SLE. An autoimmune origin of a small percentage of cases of MFI is possible but unproven. The identification of antibodies to plausible antigens, such as urokinase plasminogen activator receptor, on the syncytiotrophoblastic surface would strengthen this hypothesis21
Discordance for MFI in dichorionic twins has been reported in which both twins were live born48. I have seen this occurrence as well as cases in which the second smaller twin is stillbirth with MFI confined to his or her dichorionic placenta (Fig18).
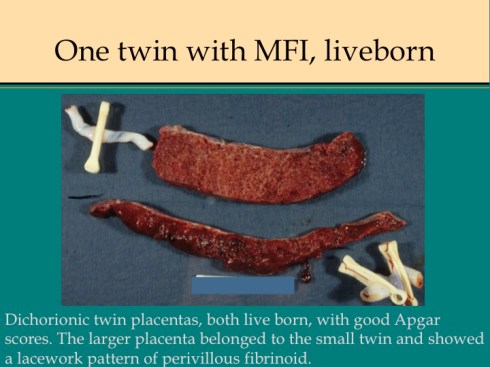
Fig 18a

Fig 18b

Fig 18c
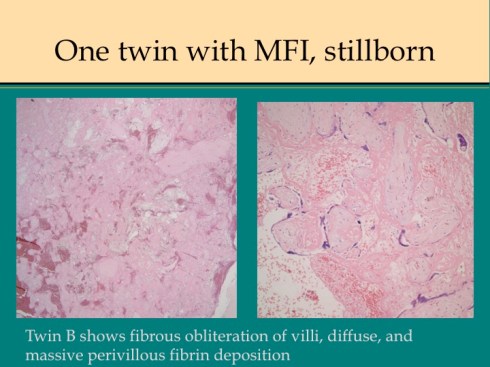
Fig 18d

Fig 18e
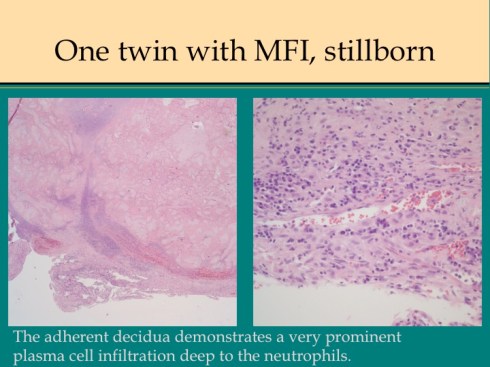
Fig 18f
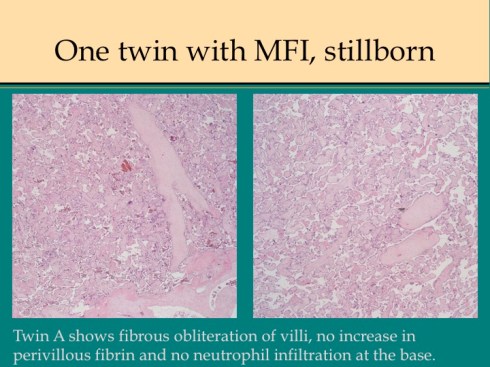
Fig 18g
This discordance for MFI should not be confused with increased intervillous fibrin that occurs with prolonged intrauterine retention following fetal death49 (Fig 19).
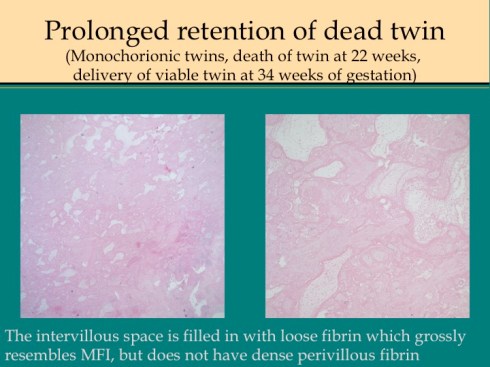
Fig 19
In my experience, stillbirth associated with maternal thrombophilia, including anti-phospholipid syndrome, demonstrates multiple placental infarctions not MFI. The only possible exception was MFI in the placenta of a mother who had multiple sickle cell crises during her pregnancy (Fig 20).
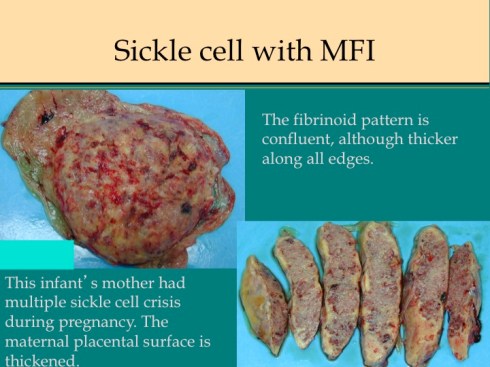
Fig 20a
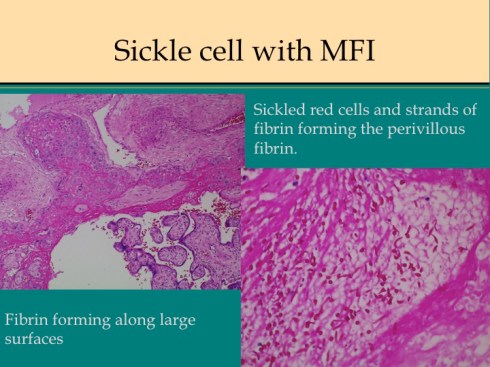
Fig 20b
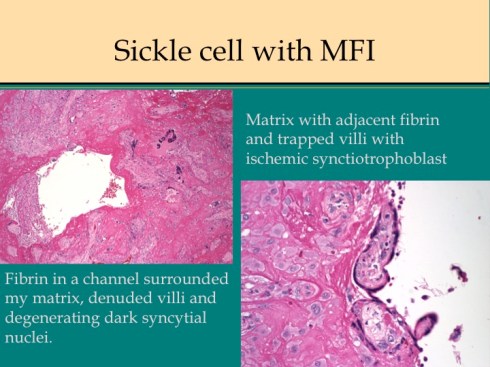
Fig 20c

Fig 20d
The lack of association of thrombophilia with MFI is surprising. Plausibly, perivillous fibrin deposition can result from turbulent intervillous blood flow causing blood stasis and ischemic syncytiotrophoblastic injury resulting in fibrin precipitation. In a hypercoagulable state, this process could become exaggerated, increasing perivillous fibrin, which in turn creates more local turbulence, further increasing fibrin deposition until MFI results. In a study of 14 patients with prior MFI, 7 of 8 treated with anticoagulation had normal weight infants compared to 2 of 6 untreated22. However, the paper does not explicitly state that the untreated mothers had recurrent MFI. In 20 women with MFI there were no single nucleotide polymorphisms associated with genes in the coagulation system34. A possible explanation for the lack of cases of MFI with thrombophilia is that the fetal losses occur in the first trimester at a time when the lesion is not well defined50, 51. Anti-phospholipid syndrome fetal loss is often in the first trimester. A more likely explanation is that the placental anti-thrombin/ fibrin systems are sufficient to overcome those thrombophilias that are compatible with maternal survival.
Bottom line:
MFI is diagnosed grossly in the placenta by finding a firm parenchymal texture due to an increased amount of perivillous fibrinoid. There may be cases that are borderline, but this definition serves well in practice. Placentas with marked maternal fibrinoid confined to the basal plate may also be in the spectrum of MFI. Recognition of the lesion is important because of the association with growth retardation and stillbirth which may reoccur in subsequent pregnancies. Early delivery of the infant may be life saving, and in some cases MFI can be recognized on prenatal ultrasound.
There is likely more than one etiology for the lesion. There are different patterns most notably a diffuse pattern versus a coarsely distributed pattern sometimes appearing to accentuate the periphery of the spiral artery flow in the intervillous space. Both patterns may become virtually confluent. The amount of matrix proliferation also varies markedly among cases. The patterns suggest specific mechanisms, but the etiology of most cases remains obscure. There are potential associations with autoimmune disease, metabolic disease, and possibly certain thrombophilias (but not the commonly diagnosed forms). MFI may occur in one of dichorionic twins.
References:
- Kaufmann P, Huppertz B, Frank HG. The fibrinoids of the human placenta: origin, composition and functional relevance. Anat Anz 1996;178:485-501.
- Sutcliffe RG, Davies M, Hunter JB, Waters JJ, Parry JE. The protein composition of the fibrinoid material at the human uteroplacental interface. Placenta 1982;3:297-308.
- Nelson DM, Crouch EC, Curran EM, Farmer DR. Trophoblast interaction with fibrin matrix. Epithelialization of perivillous fibrin deposits as a mechanism for villous repair in the human placenta. Am J Pathol 1990;136:855-65.
- Huppertz B, Kertschanska S, Frank HG, Gaus G, Funayama H, Kaufmann P. Extracellular matrix components of the placental extravillous trophoblast: immunocytochemistry and ultrastructural distribution. Histochem Cell Biol 1996;106:291-301.
- Frank HG, Huppertz B, Kertschanska S, Blanchard D, Roelcke D, Kaufmann P. Anti-adhesive glycosylation of fibronectin-like molecules in human placental matrix-type fibrinoid. Histochem Cell Biol 1995;104:317-29.
- Frank HG, Malekzadeh F, Kertschanska S, et al. Immunohistochemistry of two different types of placental fibrinoid. Acta Anat 1994;150:55-68.
- Nanaev AK, Milovanov AP, Domogatsky SP. Immunohistochemical localization of extracellular matrix in perivillous fibrinoid of normal human term placenta. Histochemistry 1993;100:341-6.
- Matejevic D, Neudeck H, Graf R, Muller T, Dietl J. Localization of hyaluronan with a hyaluronan-specific hyaluronic acid binding protein in the placenta in pre-eclampsia. Gynecol Obstet Invest 2001;52:257-9.
- Kisalus LL, Herr JC. Immunocytochemical localization of heparan sulfate proteoglycan in human decidual cell secretory bodies and placental fibrinoid. Biol Reprod 1988;39:419-30.
- Ramsey EM, Donner MW. Placental vasculature and circulation. Philadelphia: W. B. Saunders Company Ltd, 1980.
- Nelson DM, Swanson PE, Davison BB, Baskin GB, Enders AC. Ontogenetic and phylogenetic evaluation of the presence of fibrin-type fibrinoid in the villous haemochorial placenta. Placenta 1997;18:605-8.
- Farmer DR, Nelson DM. A fibrin matrix modulates the proliferation, hormone secretion and morphologic differentiation of cultured human placental trophoblast. Placenta 1992;13:163-77.
- Toki T, Horiuchi A, Ichikawa N, Mori A, Nikaido T, Fujii S. Inverse relationship between apoptosis and Bcl-2 expression in syncytiotrophoblast and fibrin-type fibrinoid in early gestation. Mol Hum Reprod 1999;5:246-51.
- Marzioni D, Muhlhauser J, Crescimanno C, Banita M, Pierleoni C, Castellucci M. BCL-2 expression in the human placenta and its correlation with fibrin deposits. Hum Reprod 1998;13:1717-22.
- Nelson DM. Apoptotic changes occur in syncytiotrophoblast of human placental villi where fibrin type fibrinoid is deposited at discontinuities in the villous trophoblast. Placenta 1996;17:387-91.
- Austgulen R, Chedwick L, Vogt Isaksen C, Vatten L, Craven C. Trophoblast apoptosis in human placenta at term as detected by expression of a cytokeratin 18 degradation product of caspase. Arch Pathol Lab Med 2002;126:1480-6.
- Smith SC, al e. Increased placental apoptosis in intrauterine growth restriction. Am J Obstet Gynecol 1997;177:1395-401.
- Ratts VS, Tao XJ, Webster CB, et al. Expression of BCL-2, BAX and BAK in the trophoblast layer of the term human placenta: a unique model of apoptosis within a syncytium. Placenta 2000;21:361-6.
- Leung DN, Smith SC, To KF, Sahota DS, Baker PN. Increased placental apoptosis in pregnancies complicated by preeclampsia. Am J Obstet Gynecol 2001;184:1249-50.
- Moe N, Jorgensen L. Fibrin deposits on the syncytium of the normal human placenta: evidence of their thrombogenic origin. Acta Pathol Microbiol Scand 1968;72:519-41.
- Pierleoni C, Castellucci M, Kaufmann P, Lund LR, Schnack Nielsen B. Urokinase receptor is up-regulated in endothelial cells and macrophages associated with fibrinoid deposits in the human placenta. Placenta 2003;24:677-85.
- Fuke Y, Aono T, Imai S, Suehara N, Fujita T, Nakayama M. Clinical significance and treatment of massive intervillous fibrin deposition associated with recurrent fetal growth retardation. Gynecol Obstet Invest 1994;38:5-9.
- Katz VL, DiTomasso J, Farmer R, Carpenter M. Activated protein C resistance associated with maternal floor infarction treated with low-molecular-weight heparin. Am J Perinatol 2002;19:273-7.
- Robins JC, Heizer A, Hardiman A, Hubert M, Handwerger S. Oxygen tension directs the differentiation pathway of human cytotrophoblast cells. Placenta 2007;28:1141-6.
- Feinberg RF, Kliman HJ, Lockwood CJ. Is oncofetal fibronectin a trophoblast glue for human implantation? Am J Pathol 1991;138:537-43.
- Fox H. Fibrinoid necrosis of placental villi. J Obstet Gynaecol Br Commonw 1968;75:448-52.
- Fox H. Perivillous fibrin deposition in the human placenta. Am J Obstet Gynecol 1967;98:245-51.
- Andres RL, Kuyper W, Resnik R, Piacquadio KM, Benirschke K. The association of maternal floor infarction of the placenta with adverse perinatal outcome. Am J Obstet Gynecol 1990;163:935-8.
- Katzman PJ, Genest DR. Maternal floor infarction and massive perivillous fibrin deposition: histological definitions, association with intrauterine fetal growth restriction, and risk of recurrence. Pediatr Dev Pathol 2002;5:159-64.
- Kaufmann P. Experiments on infarct genesis caused by blockage of carbohydrate metabolism in guinea pig placentae. Virchows Arch A Pathol Anat Histol 1975;368:11-21.
- Matern D, Schehata BM, Shekhawa P, Strauss AW, Bennett MJ, Rinaldo P. Placental floor infarction complicating the pregnancy of a fetus with long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency. Mol Genet Metab 2001;72:265-8.
- Carruth A, Strauss A, Bennett M, Narayan S, Ernst L. Mutations in Long-Chain 3-Hydroxyacyl-CoA Dehydrogenase and Placental Maternal Floor Infarct/Massive Perivillous Fibrin DepositionSociety for Pediatric Pathology Interim Meeting. Philadelphia, PA, 2009.
- Mandsager NT, Bendon R, Mostello D, Rosenn B, Miodovnik M, Siddiqi TA. Maternal floor infarction of the placenta: prenatal diagnosis and clinical significance. Obstet Gynecol 1994;83:750-4.
- Uxa R, Baczyk D, Kingdom JC, Viero S, Casper R, Keating S. Genetic polymorphisms in the fibrinolytic system of placentas with massive perivillous fibrin deposition. Placenta 2010;31:499-505.
- Brown DL, DiSalvo DN, Frates MC, Davidson KM, Genest DR. Placental surface cysts detected on sonography: histologic and clinical correlation. J Ultrasound Med 2002;21:641-6; quiz 647-8.
- Wagner J, Hustin J, Bonno M, Kephart G, Gurian K, Gleich G. Pregnancy-associated major basic protein: deposition of protein and expression of mRNA at the maternal-fetal junction in early and late gestation. Placenta 1994;15:625-640.
- Wasmoen T, Coulam C, Benirschke K, Gleich G. Association of Immunoreactive Eosinophil Major Basic Protein with Placental Septa and Cysts. American Journal of Obstetrics and Gynecology 1991;165:416-420.
- Vernof KK, Benirschke K, Kephart GM, Wasmoen TL, Gleich GJ. Maternal floor infarction: relationship to X cells, major basic protein, and adverse perinatal outcome. Am J Obstet Gynecol 1992;167:1355-63.
- Naeye RL. Maternal floor infarction. Hum Pathol 1985;16:823-8.
- Bane AL, Gillan JE. Massive perivillous fibrinoid causing recurrent placental failure. Bjog 2003;110:292-5.
- Clewell WH, Manchester DK. Recurrent maternal floor infarction: a preventable cause of fetal death. Am J Obstet Gynecol 1983;147:346-7.
- Bendon RW, Hommel AB. Maternal floor infarction in autoimmune disease: two cases. Pediatr Pathol Lab Med 1996;16:293-7.
- Silva CA, Sultan SM, Isenberg DA. Pregnancy outcome in adult-onset idiopathic inflammatory myopathy. Rheumatology (Oxford) 2003;42:1168-72.
- Al-Adnani M, Kiho L, Scheimberg I. Recurrent placental massive perivillous fibrin deposition associated with polymyositis: a case report and review of the literature. Pediatr Dev Pathol 2008;11:226-9.
- Satoh M, Ajmani AK, Hirakata M, Suwa A, Winfield JB, Reeves WH. Onset of polymyositis with autoantibodies to threonyl-tRNA synthetase during pregnancy. J Rheumatol 1994;21:1564-6.
- Hung NA, Jackson C, Nicholson M, Highton J. Pregnancy-related polymyositis and massive perivillous fibrin deposition in the placenta: are they pathogenetically related? Arthritis Rheum 2006;55:154-6.
- Sebire NJ, Backos M, Goldin RD, Regan L. Placental massive perivillous fibrin deposition associated with antiphospholipid antibody syndrome. Bjog 2002;109:570-3.
- Redline RW, Jiang JG, Shah D. Discordancy for maternal floor infarction in dizygotic twin placentas. Hum Pathol 2003;34:822-4.
- Ashworth C, Stouffer J. A study of fibrin deposition in the placenta. Am J Clin Pathol 1956;26:1031-1043.
- Rushton D. The classification and mechanisms of spontaneous abortion. Perspect Pediatr Pathol 1984;8:269-287.
- Svensson AM, Waters BL, Laszik ZG, et al. The protein C system in placental massive perivillous fibrin deposition. Blood Coagul Fibrinolysis 2004;15:491-5.
