What is villitis?
The name simply implies inflammation that occurs in the placental villi. This broad concept is subcategorized into pathologic entities. These subcategories for villitis were first established by Altshuler and Russell 1. Their category of diffuse villitis, which was described primarily as villous edema and immaturity, and is not usually considered villitis today. Their category of acute villitis is also not usually considered villitis as much as a rare component of ascending infection and will be considered in that chapter, but the appearance at low magnification can mimic chronic villitis

Fig 1) At low magnification, there appears to be a nest of typical chronic villitis. (H&E, 10x)

Fig 2) At high magnification, the villitis demonstrates a high percentage of neutophils consistent with acute villitis. (H&E 40x)
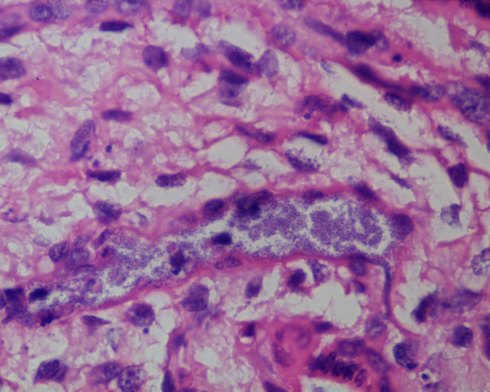
Fig 3) Under oil immersion numerous cocci are present in this second trimester fetal loss from Group B beta-hemolytic Streptococcus infection. (H&E, 100x)
(Fig 1-3). Villous edema and acute inflammation may be a minor component of other categories of chronic villitis.
The major category of chronic villitis is lympho-histiocytic villitis that subsumes Altshuler and Russell’s proliferative and focal villitis. The key features are an infiltration of lymphocytes and destruction of the capillary bed within villi. They described variants lymphohistiocytic villits with necrosis, reparative changes that could have granulomatous components and a fibrotic variant (now called avascular villi). They suggested that these were progressive from acute to scarred stages of lymphohistiocyic villitis.
Within chronic villitis there is a major etiologic division into infectious villitis versus villitis of unknown etiology (VUE). The proof that a placenta with chronic lymphohistiocytic villitis is VUE and not infectious villits, is one of exclusion. In VUE there is no evidence of infection in the placenta or in the infant. In one study, the cord blood in placentas with presumed VUE did not have elevated IgM, except for one case with plasma cells2. The presence of plasma cells is considered atypical of lympho-histiocytic villitis, and perhaps should be named plasma cell villitis. A rare plasma cell may be found in VUE, but plasma cells are often a marker of intravillous infection and their presence requires active exclusion of an infectious etiology. Villitis due to infection will be considered in the chapter on blood borne infection in the fetus.
The pathogenesis of VUE
The lymphocytic inflammatory destruction of small vessels in VUE reminded observers of the changes seen in transplant rejection. Other features were consistent with this analogy. The inflammatory cells most commonly found in VUE are T lymphocytes and activated macrophages 3,4. Dr. Faulk and collaborators using fluorescent immunohistochemical study of 25 otherwise normal placental frozen sections found villitis associated not only with T helper cells (CD4) and activated macrophages (HLA-DR, HLA-DP, HLA-DQ), but basement membrane deposition in trophoblast and fetal endothelium of factor IX, and of platelets, and of loss of von Willebrand immunostaining in stem vessels4. Another immuno-localization study found that the T-cells expressed T regulatory markers CD25 and Fox P35. To determine the origin of these T-lymphocytes Redline and Patterson used in situ hybridization of X and Y chromosome probes in three male placentas and demonstrated that the T cells in villitis were maternal6. Similar results were obtained in a single placenta from a stillborn male fetus with severe villitis using a technique that combined in situ identification of X and Y chromosomes in formalin fixed tissue with immunohistochemistry of antigens for macrophages (CD-68), pan T cell (CD3) and cytokeratin7. The T cells in the villitis were 90% female, but the macrophages were only 10% female, the remaining male macrophages were likely to be intrinsic fetal Hofbrauer cells. Hofbrauer macrophages are part of the normal villous cell population and are not per se evidence of inflammation. Of note, pervillous macrophages were as expected maternal, and areas of villitis often had gaps in the syncytiotrophoblast covering as demonstrated by AE1/AE3 cytokeratin immunostain.
A different approach confirmed that the T lymphocytes were maternal. A study of 3 frozen sections of placenta, 1x1x.5 cm taken near the base from 3 uncomplicated pregnancies and from 4 with intrauterine growth retardation immuno-stained with a monoclonal antibody to HLA-DRw 52 for which mother and fetus were discordant. The study found that the percentage of HLA-DR positive cells in villi varied from 75-100% maternal8. In the uncomplicated pregnancies only 0.6 to 1.5% of villi had HLA-DR positive cells, but in those with IUGR, 10-28% of villi were involved. These incidences are likely high because of the sensitivity of the method compared to a pathologic assessment of the diagnosis based on multiple inflammatory cells and loss of capillaries to diagnose villitis. The authors point out that having some maternal inflammatory cells may be normal, but this does not prove that those cells are able to enter the fetal circulation.
These observations support a widely accepted hypothesis that VUE is a focal area of maternal T cell rejection of fetal cells expressing paternal histocompatibility antigens. Normally these fetal cells are not detected by maternal T-lymphocytes if they are within the intact syncytium, which does not express allogenic histocompatibility antigens. Some of the T-lymphocyte cytokines could lead to necrosis and migration of neutrophils. Other cytokines from T cells and macrophages may direct the subsequent organization and scarring of the damaged villous stroma, and even initiate a granulomatous response. This rejection hypothesis also explains the observation of intravascular thrombosis and of vascular destruction. The up-regulation of thrombogenic substances is part of the rejection process, which would favor fetal vascular thrombi.
What initiates VUE?
The initiating cause of villitis has always been perplexing. Russel cites a personal communication with Dr. William Blanc that proposed that VUE was a form of graft-versus-host rejection9. Dr. Russell on the other hand still thought the inflammation was predominantly fetal in origin, and that this observation favored infection. Even if the facts of cell origin proved wrong, the two theories remain the contenders for the cause of VUE. The idea of a primary immune rejection must answer what initiates the process in only some infants. The suggestion of infection initiating the process must contend with the lack of a proven organism or of disease in the newborn.
Jacques and Qureshi found asymmetry of VUE between twins, both in degree and presence, but without a statistical difference in birth weight10. The dichorionic differences would favor a primary immunologic basis in that T-cells may recognize only paternal antigens in one of two genotypes. However, the clarity of this was rebutted in the same study in that a monochorionic twin placenta was also asymmetric for VUE.
A case report of neonatal alloimmune thrombocytopenia that also demonstrated IgG HLA class I antibodies. The placenta demonstrated VUE with fetal thrombotic vasculopathy (hemorrhagic endovasculosis and microthrombi)11.
The best documented experimental studies of transplacental passage of maternal T cells into the fetus were done using rat T cells already activated against a different strain of rat and then injected into the pregnant mother12-14. Depending on the dose, a percentage of the infant rats developed severe growth retardation and death due to engrafted maternal cells producing a systemic graft versus host reaction. However, the rat placenta has a different morphology, and the studies did not report placental pathology. However, the experiment does demonstrate that such T-cells can pass to the fetal rat, presumably through the placenta. In human patients, it is unclear how often mothers with VUE in their placenta have T cells activated to paternal antigens in their circuation.
Immunolocalization studies provide some possible insight into causation. The finding of immunoregulatory T-cells perhaps explains why VUE is often localized and does not spread throughout the placenta. If this is true, then each new focus represents a new process. The same immuno-localization study found evidence of phosphorylation of the STAT-1 in syncytiotrophoblast in the region of VUE5. This P-STAT-1 is normally translocated to the nucleus and activates interferon gamma. They suggest that the trophoblast itself calls up the inflammatory response. Given the scattered repetitive pattern of some VUE, the findings are compatible with a response to syncytiotrophoblast infection with a virus. It is possible that the resulting VUE is protective of the fetus. Of course this is all speculation without more detailed understanding.
What do we see from routine examination of the placenta with lympho-histiocytic villitis of unknown etiology (VUE)?
Gross: The most common gross lesion is to not be visible which is consistent with the scale of the usual scattered small lesions. Less frequently the lesion shows pale villi that are not agglutinated but showing a normal granular placental texture (Fig 4)
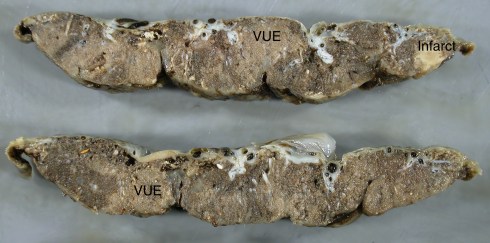
Fig 4) These cut sections of fixed placenta demonstrate many pale areas of extensive VUE.
. The lesions appear contiguous and consistent with involvement of larger proximal villous structures extending outward into the distal villous tree. Rarely a focus of VUE shows a coarse granularity similar to that in massive pervillous fibrinoid (Fig 5).

The is a placenta with coarsely granular villi especially in the middle slice that demonstrated extensive VUE.
In the majority of placentas with VUE no gross abnormality is recognized with routine pathological examination.
Microscopic:
At low magnification, VUE demonstrates villi that are usually plumper than surrounding non-inflamed villi, show a loss of capillaries, and have a blue cast from the infiltrating lymphocytes and activated macrophages (Fig 6).

Fig 6) At low magnification, the area of the slide on the left shows larger, more basophilic villi typical of VUE. (H&E,4x)
A similar low magnification pattern can be seen with distal fetal vascular occlusive lesions, but the histologic features are distinct at higher magnification. In VUE, the inflammatory cells are the predominant feature and are primarily monocytic although neutrophils and even eosinophils may occasionally be present. (Fig 7,8).

Fig 7) At medium magnification, there is VUE within the anchoring villus at the maternal floor (basal villitis). There is a small infiltrate in continuity with both the villus and the maternal floor that at the arrows shows eosinophlis. (H&E, 20x)

Fig 8) At higher magnification binucleate eosinophils can be identified (arrows) (H&E, 40x)
In practice, there is a wide range of pathological change present in VUE that includes the necrosis and shedding of syncytiotrophoblast, the deposition of fibrin and fibrinoid, the necrosis of villous structures with some neutrophils, inflammatory giant cells, intervillous monocytes, and ongoing villous, sometime granulomatous, repair. These features are all well described and illustrated in the early paper by Russell9. There is no single feature on H&E pathology that is unequivocally more significant clinically than another. This complexity can be appreciated by looking at two representative cases. Case 1: The mother was a primigravida. The infant was AGA, delivered at term with Apgars of 9/9. The placenta was appropriate size with a 7 cm subchorionic cyst and no other gross lesions (Fig 9-13).

Fig 9) At low power, extending radially (lines) from a septal cyst is an area of thick villi with a “haze” around the edges. The surrounding villi are very mature with many small terminal villi. In the center of the abnormal area there is a small “nidus” of agglutinated eosinophilic villi (*). (The septal cyst is more of a cytotrophoblast island surrounded by fibrinoid and adherent villi, and any relationship of the area of VUE to a septum cannot be determined from the slide) (H&E 2x)

Fig 10) At medium power, this image demonstrates typical features of VUE. The interior of the large villus in the center shows a loss of identifiable capillaries, and an intense infiltration of lymphocytes. Another prominent feature is the necrosis of the syncytiotrophoblast (arrows on some examples). A small villus nearby (*) is a useful comparison. (H&E, 20x)
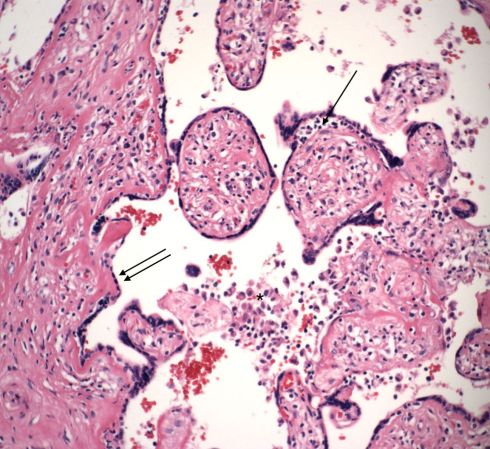
Fig 11) The single arrow demonstrates a collection of lymphcytes separating the syncytiotrophoblast from the underlying stroma. The double arrows demonstrate the appearance of syncytiotrophoblast over an area of fibrinoid, likely a repair similar to re-epithelialization. The * is over a small collection of intervillous monocytes. (H&E, 20x)

Fig 12) The villi in the center appear denuded of syncytiotrophoblast and are becoming agglutinated with fibrin. While a certain identification of the cells with larger nuclei (arrows) cannot be made, they appear to be cytotrophoblast that will contribute the secreted proteins to the fibrin to create fibrinoid. (H&E, 20x)

Fig 13) The villi in this image are adjacent to villi with VUE. The center villous is likely a mature intermediate villsu, but overall there appears to be an increase in capillaries from dilatation from diverted blood flow from an a connected stem villous or possibly from vascular growth factors generated by the VUE. (H&E, 20x)
Case 2: This placenta was from an AGA term infant. Fifty percent of the placental villi were affected with VUE, and the placenta parenchyma had an abnormal granularity (Fig 14-18).

Fig 14) At low magnification, there is distinct cluster (oval)of fibrinoid agglutinated, avascular villi. Many foci in the remaining areas show larger, basophilic villi due to VUE, compared to a smaller number of areas showing distribution of terminal villi in a mature placenta. (H&E, 2X)

Fig 15) At intermediate magnification, a smaller area of agglutinated mostly avascular villi (oval) can be seen. The surrounding villi all demonstrate VUE with a variety of features including occluded vessels (V), a collection of lymphocytes beneath the syncytium (S), and collections of intervillous monocytes (IV). There are some syncytial knots (K). While these knots could be residual of normal maturation, it is not unusual to see knots from syncytial apoptosis in areas of ischemia, but preservation of the cytotrophoblast in other conditions of perivillous fibrinoid formation. (H&E, 10X)
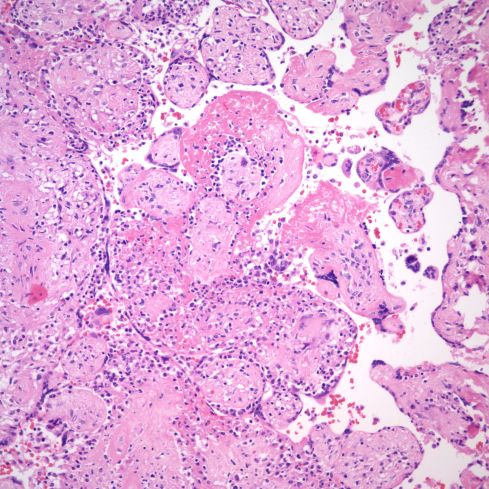
Fig 16) This image shows similar features to the previous with subsyncytial lymphocytes, syncytial necrosis and perivillous fibrin forming around avascular villi with some lymphocytes but also reparative changes that will end in scarring. (H&E, 20x)

Fig 17) This area shows a more reparative process in the villi at the top, and a florid destructive process in the villi near the bottom of the image. (H&E,20x).

Fig 18) At high magnification of a villous that appears to be exploded by villitis there are neutrophils (arrow) and necrotic debris, as well as macrophage giant cells. (H&E, 40x)
Fetal Vascular Thrombi in VUE
There are a few histological observations that will be considered in more detail. The first is the presence of thrombi and fetal thrombotic occlusive changes.
Small thrombi are sometimes seen in the capillaries (Fig 19).

Fig 19) This terminal villus with VUE shows a small fibrin thrombus in a capillary (arrow). (H&E, 40x)
There is loss of identifiable capillaries and the villi show a progression to scarred, avascular villi (Fig 20, 21).

Fig 20) This intermediate magnification image shows a visually plausible a progression from VUE to avascular villi. (H&E, 20x)
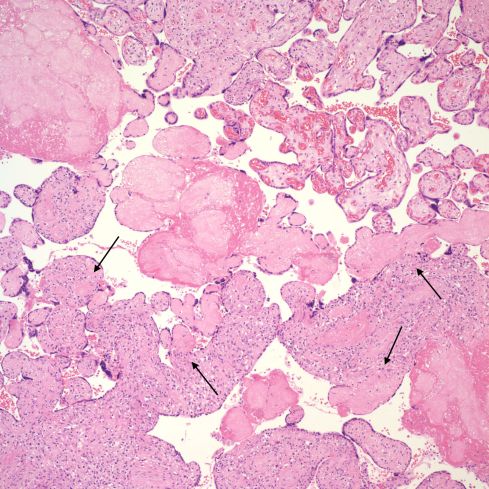
Fig 21 This intermediate magnification shows a band of avascular villi cutting diagonally across the center of the field. There are also avascular villi that appear attached to villi with VUE (arrows). These small avascular villi often still have syncytiotrophoblast over the surface. They may all be the result of a collision of two separate events, with an early fetal vascular occlusion and a more recent VUE, or they may show progression from VUE to avascular villi. Even if they are separate events the avascular villi could still be a consequence of earlier VUE.
VUE is not restricted to terminal and intermediate villi, but can appear in larger stem villi (Fig 22,23).

Fig 22) There is chronic, somewhat granulomatous inflammation in this stem villus that appears primarily vasocentric with loss of the media seen well in the lower portion of the field between the arrows. The blood in the lumen demonstrates some basophilia and repair around the margins suggestive of vascular stasis. (H&E, 20x)
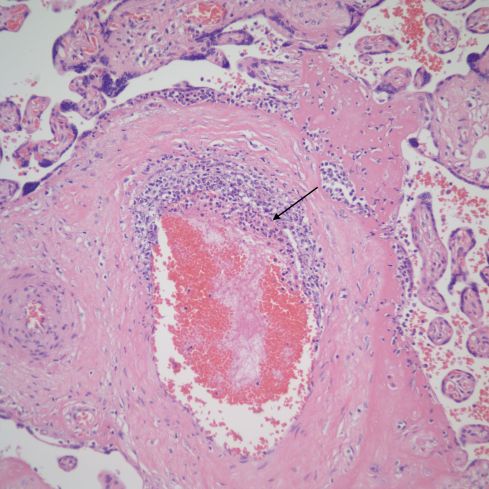
Fig 23) This is another example of a stem villus with vasocentric as well as subsyncytial inflammation. Note that the vascular inflammation is in the region of the subsyncytial inflammation. There is early lumenal fibrin deposition (arrow).
These lesions of larger stem villi may show endovascular lymphocytes and thrombus (Fig 24-26).

Fig 24) The stem villus shows eccentric inflammation and destruction of a portion of the vessel wall, and fibrin deposition. In the upper left corner the connecting and adjacent villi show VUE and the syncytial surface of the stem villus shows necrosis and lymphocytic inflammation. (H&E, 10x)
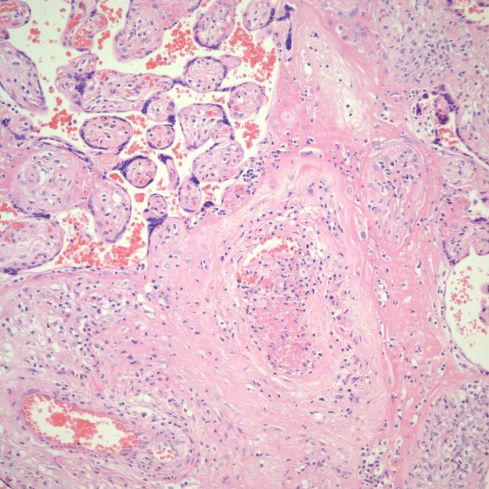
Fig 25)This stem vessel in an area of villitis shows lumenal thrombus and inflammation of the vessel wall. (H&E, 20x)

Fig 26 There is a large, organizing thrombus in a stem villous vessel with VUE in the adjacent and connected villi. (H&E, 20x)
Diagnostically, the pathologist usually tries to separate a primary fetal thrombotic vasculopathy from up-regulation of thrombogenesis in VUE. There are no definitive criteria, but finding fetal thromboitic lesions in isolation of VUE in some areas suggests a primary thrombotic process. Finding focal thrombi within areas of VUE, especially in capillaries, favors VUE as the primary process.
Perivillous fibrinoid and VUE
The second important observation is that VUE has been associated with increased perivillous fibrinoid. The relationship of cause and effect on a slide can be difficult. Two lesions may be associated but may be just a collision of two different but common processes. However, the fibrinoid deposition with villitis at least demonstrates a plausible pathogenesis as a consequence of VUE as seen in the figures from the two cases studies above. The fibrinoid is often present surrounding a single villus, but also may form an enmeshed nidus of villi (Fig 27-32).

Fig 27) A terminal branch from an intermediate villus with VUE has lost its syncytiotrophoblast and fibrin has linked it to surrounding scarred vill between the asterisks. The arrows point to likely cytotrophoblasts that will add their secretions (fetal fibronectin, annexins) to the fibrinous fibrinoid. (H&E, 20x)
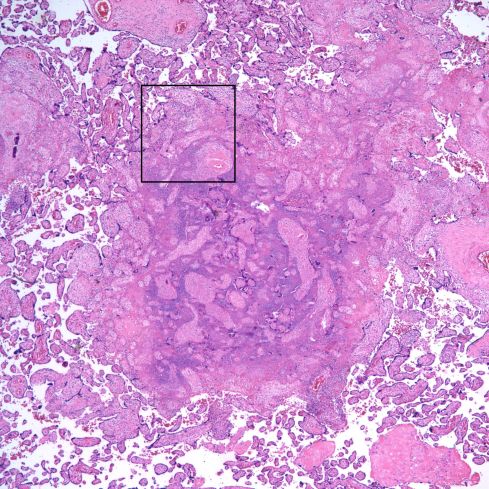
Fig 28) There is core of villi enmeshed in a basophilic matrix surrounded by villi demonstrating VUE. The rectangle is shown at higher magnification in the next figure. (H&E, 4X)
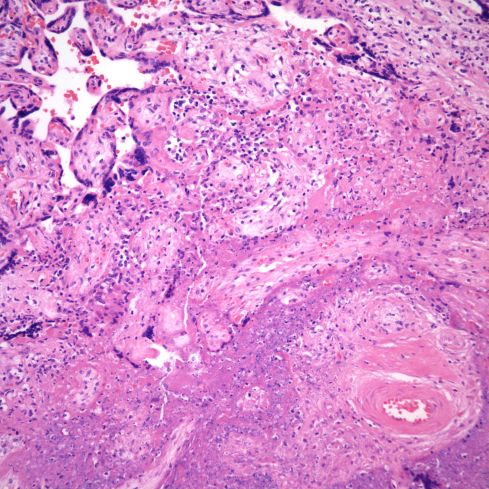
Fig 29) At higher magnification, the basophilic matrix can be seen to be similar to the fibrinous fibrinoid matrix of perivillous fibrinoid deposition with the addition of numerous inflammatory nuclei from perivillus and subsyncytial inflammatory cells. (H&E, 20x)

Fig 30) Another similar focus of enmeshed villi with VUE in another area of the same placenta as figure 5609. (H&E, 4x)

Fig 31) At low magnification, this focus of enmeshed villi in VUE has a more granular appearance of the basophilic material between villi (*), than typical in such nests. (H&E, 10x)

Fig 32) At higher magnification, the blue material shows extensive cell debris and neutrophils (arrows). (H&E, 40x)
As discussed in the chapter 10b on massive perivillous fibrinoid infiltration (MFI), the origin of this fibrinoid is syncytiotrophoblastic injury. If the syncytiotrophoblast is lost over the surface of the villus 1) connective tissue is exposed that will favor fibrin formation from maternal blood, and 2) viable cytotrophoblast will secrete a fibrinoid of sticky proteins such as fibronectin and annexins. Syncytiotrophoblast depends on the maternal circulation for it oxygen, and would not be expected to perish from the loss of fetal circulation. In VUE lymphocytes are commonly concentrated just beneath the syncytium, and the syncytiotrophoblast often appears necrotic and/or lifting off the villus. Plausibly cytokines from the T-cell response even if not directed at the trophoblast might still cause cell damage. Microscopically, VUE and perivillous fibrinoid can occur without the other; thus, there is no necessary pathogenic relationship. VUE may or may not have syncytial necrosis and fibrinoid, and pervivillous fibrinoid may form without VUE.
The associated fibrinoid and syncytial necrosis may be associated with clusters of intervillous macrophages, which as noted above are maternal. These clusters have been designated chronic intervillositis, and this entity will be addressed in a later section.
Basal Villitis
Another distinctive histologic feature in some cases VUE is that the lesion is confined to or predominant in the basal villi, referred to as basal villitis. These basal villi at the point of contact do not have an intervening syncytiotrophoblast layer (Fig 33-36).

Fig 33) This low magnification demonstrates an anchoring villus that is directly embedded (*) in the maternal floor cytotrophoblast layer (MF). The small branches near the point of contact show VUE in continuity with inflammation at the point of contact (arrows). The area in the rectangle is enlarged in the next figure. (H&E, 4x)
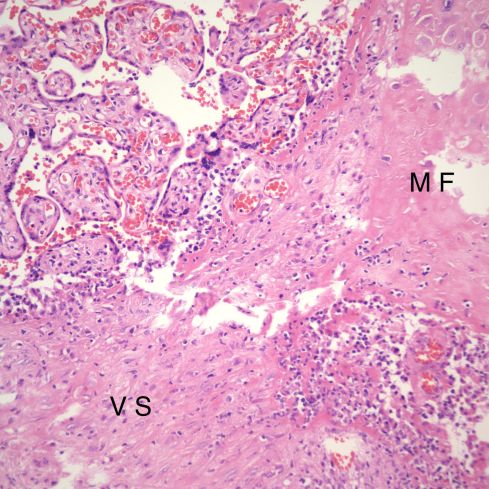
Fig 34) The villous stroma (V S) is directly embedded in the maternal floor (M F). The edges of the villi show loss of syncytiotrophoblast, fibrin, and lymphocytic infiltration. (H&E, 20x)
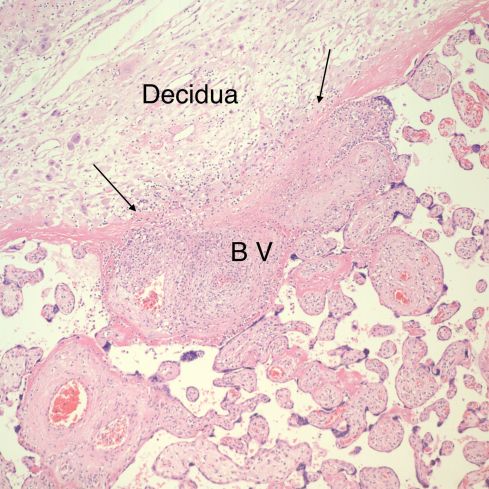
Fig 35) The basal villus (B V) demonstrates a loss of the fibrinoid layer (between the arrows) and some disruption of the cytotrophoblastic shell at the point of contact. There is a concentration of lymphocytes in the decidua beneath the contact and in continuity with the inflammation in villus. (H&E, 10x)
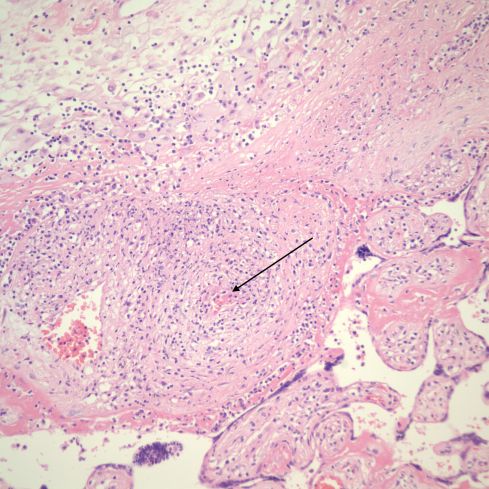
Fig 36) A higher magnification of the prior figure showing more clearly the lymphocytes appearing to percolate through the cytotrophoblastic shell of the maternal floor. The arrow demonstrates a small thrombus in a the anchoring villus vessel. (H&E, 20x)
Inflamed villi adjacent to maternal septa are also considered to be basal villitis as they also are in contact with the same components of the maternal floor (fig 37,38).
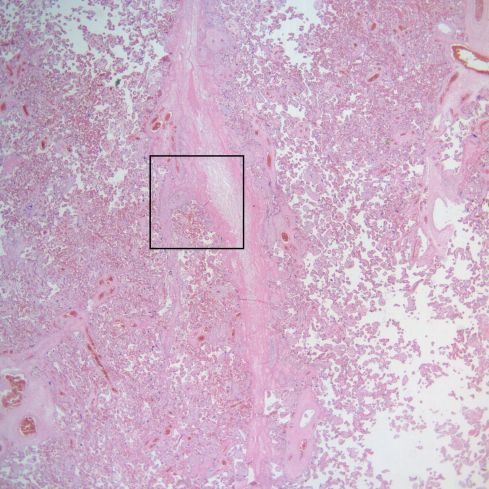
Fig 37) This low power image shows a linear maternal septum running down the center. The rectangular area is enlarged in the next figure. (H&E, 2x)

Fig 38) The higher power demonstrates VUE in villi in contact with the septum. (H&E, 10x)
Labarre and Faulk demonstrated the same immunologic phenotype in the basal villi as in the non-anchoring villi with VUE15. Plausibly the etiology of basal villitis could be different from VUE in the parenchymal villi. T-lymphocytes may need a break in the syncytium to enter villi in the parenchyma, but in the basal villi there may be a direct path through the chorionic shell from the decidua (Fig 39,40).

Fig 39) This low magnification of the maternal floor shows a large area of VUE that is in continuity with villi embedded in the maternal floor in areas with small gaps in the chorionic shell. (arrows) (H&E, 10x)

Fig 40) This medium magnification shows a focus of VUE between the arrows embedded in the maternal floor in a gap in the chorionic shell. There are still some strands of fibrinoid. The lymphocytes in the decidua in this area, imaginatively, are streaming into the villi. (H&E, 10x)
Beside the maternal floor, another location that maternal blood is exposed to at least a few fetal stromal cells is the subchorionic plate. In this location, as gestation progresses, the lining cytotrophoblast layer is usually replaced by fibrinoid, but focally this surface could be denuded. In some placentas there is a line of lymphocytes in this subchorionic inner surface of the fetal surface (Fig 41).

Fig 41) The band of blue chronic inflammation runs along the inner fetal surface of the placenta. This area is usually separated from maternal blood by either a cytotrophoblast layer, or more usually in later pregnancy by a band of fibrinoid. In this case, there is a layer of subchorionic fibrin against the stromal surface. The arrow shows the junction with the chorionic stroma without any fibrinoid barrier and possibly placental stroma had been exposed to maternal blood. (H&E, 20x)
In my experience, placentas demonstrating this inflammatory layer usually have VUE, but I have not systematically studied this association. Russell noted an association of lymphocytes in the connective tissue of the reflected chorion, which is another location in which paternal antigens could be exposed to maternal blood9. This is now called chronic chorioamnionitis, and will be discussed with membrane lesions.
Other associated lesions:
Villi adjacent to VUE may show capillary hypervascularity (42,43).

Fig 42) At low magnification, the villi show a mix of normal, VUE (*) and vascular hyperplasia (rectangle). (H&E 4x)

Fig 43) At higher magnification within the area of the rectangle, the villi demonstrate marked capillary hyperplasia. (H&E, 20x)
In theory this proliferation could be due to inflammatory mediators favoring neogenesis of capillaries from nearby inflamed villi that are somewhere in continuity. Alternatively, obstructed blood flow in one part of the tree, may be diverted to other villi and stimulate vasculogenesis.
An infrequent by plausible associated lesion is intervillous thrombus. Based on some evidence, these lesions may be due to fetal hemorrhage into the intervillous space. With VUE, if the spatial and timing were to injure the syncytiotrophoblast and to injure a fetal hemorrhage, an intervillous hemorrhage could occur (Fig 44,45).

Fig 44) The intervillous thrombus is in the lower left part of the field. A line of villi with VUE appear to stream from it (arrows). A possible explanation is a hemorrhage from a villus with VUE. (H&E, 2x)

Fig 45) At slightly higher magnification villi with VUE and avascular villi can be seen within the periphery of the intervillous thrombus. (H&E, 4x).
The Extent of VUE in a placenta
VUE often demonstrates a scattered pattern similar to that seen in infectious villitis. This pattern suggests random hits from immune cells or infectious particles in the maternal blood flow. VUE may also appear to follow the branching of a stem villous (Fig 46,47).
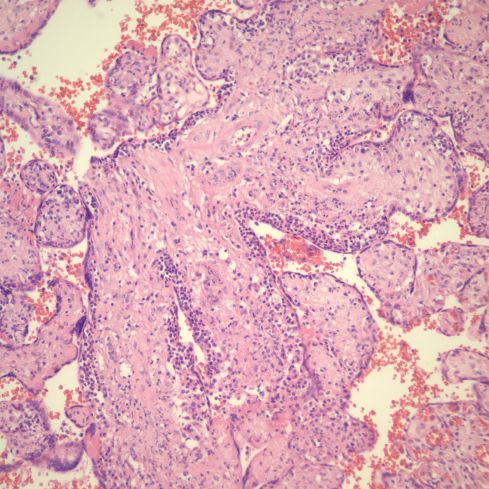
Fig 46) This is an image of a branch point of a small stem villus demonstrating how the entire branching structure is involved. Note the prominence of the subsyncytial inflammation. (H&E 20x)

Fig 47) There is a partial thrombus in a main vessel of a small stem villus. The inflammation from VUE appears to be continuous with villi branching directly from it in the plane of section, and surrounded by other villi with VUE. (H&E 20x)
A static histological slide cannot determine if the inflammation is ascending or descending in the villous tree, but does suggest that VUE in some cases can expand within the villous tree. The pattern and extent of VUE may have diagnostic significance. Certainly, the percentage of villi involved appears to correlate with some clinical complications. Quantifying the extent of villous involvement in VUE is complicated since VUE is usually only detected after microscopic sectioning. If the process is not uniformly distributed, which it usually is not, then sampling may not be representative. Nevertheless, pathologists have attempted to standardize the criteria for determining extent of VUE in a placenta. Knox and Fox proposed a simple grading system for the extent of villitis. 16.
Grade 1: 1-2 foci inflammation in 4 slides, with each focus with only a few villi involved
Grade 2: up to 6 foci, with up to 20 villi/focus
Grade 3: multiple foci, up to ½ of a low power field
Grade 4: large areas of most sections
Empirically they found that 4 sections were required to have a reliable sample. Unfortunately, routine pathology practice samples fewer than 4. A simpler but similar system was proposed by Russell that is applicable to usual practice: low: no more than 1 focus per low power field, moderate: frequent foci up to 25% of the field, and severe: more than 25%9.
What is the incidence of VUE?
Most villitis is mild and is present in approximately 5-20% of placentas without obvious harm to the fetus. Studies vary in the incidence likely due to their extant of sampling, which will increase the apparent incidence in a focal lesion, or in their histologic definition of the lesion 16,17. A unique approach to determining incidence of villitis used by Labarrere et. al. was via immuno-localization of HLA-DR positive macrophages18. They examined a total of 13.4 blocks per case (25 cases) including H&E and immuno-stained. They found villitis in 19 of 25 cases (76%), but even looking at 2.31 blocks per case, they still had 10/25 (40%) positive. This study suggests that the true incidence of VUE is higher than the incidence detected by more routine sampling.
Is there an association with fetal death or fetal growth restriction?
Growth Restriction:
The most consistent effect of VUE in multiple papers is a correlation of extensive villitis with an increased risk of small fetal size2,9,19-22. The Knox and Fox report demonstrated an incidence of VUE of136/1000 random placentas16. The only clinical correlation was with intrauterine growth restriction in their grades 2-4 of villitis. A study by Mortimer et al. of 120 consecutive placentas found 10 with villitis, of which six cases had fetal growth restriction, with 5 showing moderate and one mild villitis 2. In another study confirming a correlation of villitis with IUGR, the authors noted a significant association of villitis with normal ponderal index and speculated that this implied an early onset of the factors causing growth retardation19. The question of time of onset of VUE and duration are not usually considered in most studies. As with any association, it is difficult to determine causation. Possibly, utero-placental ischemia or subclinical infection could damage syncytiotrophoblast allowing maternal T cells into the villi, and the VUE is a secondary effect. If the extent of VUE alone with its loss of functioning villi were the cause of growth restriction, then a higher portion of infants with extensive villitis should have growth restriction. Perhaps an index of the total mass of uninvolved tertiary villi would distinguish those without growth restriction from those with it.
Stillbirth:
A meta-analysis found only 4 papers that specifically examined the relationship of villitis to stillbirth23. The authors concluded that there were not enough studies to definitively imply a relationship of VUE with stillbirth.
A study of 750 stillbirths attributed 4 deaths to villitis of unknown etiology but did not describe them in further detail24. An earlier study had attributed death to chronic villitis in 4 of 89 cases, but this may included infectious etiologies as well25. Another study comparing intrauterine growth retardation, unexplained stillbirth, and normal placentas found an increased incidence of chronic villitis in both stillbirths and growth retarded infants compared to controls, but provided no details of the individual findings26.
A case report of a gravida 5 para 5 mother who had 4 third trimester fetal deaths and one live born as 37 weeks of gestation, but with severe growth restriction, birth weight 1,790 g27. The placentas from the last 3 fetal losses and the live born infant were examined and all demonstrated severe villitis without evidence of infection in any of the infants. The stillborn infants were 38 weeks, 2575 g, 32 weeks 1,125 g, 31 weeks 980 g, and 28 weeks 970 grams. A case study of villitis in the placenta in passing relates that the infant was small for gestation, 980 grams at 34 weeks of gestation, and that the mother had within the past year delivered an earlier stillborn infant with severe villitis of the placenta7.
The mechanism of growth restriction and/or stillbirth in the infant with placental VUE has not been definitively determined. The simplest explanation is that there is a relatively acute (that is without time for placental growth in compensation) loss of villous volume that results either in loss of nutrient exchange or ultimately loss of sufficient respiratory area. Another hypothesis is that cytokines from the villitis have a growth limiting effect on the fetus. Yet another possibility is that the vascular rejection occurring in the villi has extended into the fetus. In the rat, injection of allogenic stimulated T lymphocytes from allogenic skin grafts into pregnant rats causes fetal growth restriction and death.
In the stillborn infants that I have autopsied who died without other cause but had severe villitis there was no evidence of tissue injury other than that in the placenta. This is some evidence in support of the first hypothesis that the mechanism of death is a loss of functioning placental volume.
Dr. Potenziani-Pradella (a pathology resident at the time) and I reviewed the four autopsies in my database demonstrating severe villitis and no other cause of death28. We did not find vasculitis in the fetal body, graft versus host disease or developmental lymphoid abnormality beyond thymic involution. The autolysis in the cases made it impossible to be certain that there was no mild increased apoptosis or abnormal lymphocyte infiltration in target tissues, but there were no changes severe enough to distort the basic histology of any organs. The infants, as expected, were small for gestation. Two infants had normal ultrasound and clinical findings in the weeks prior to delivery (cases 1 and 4 in table below). These finding suggest a very rapid onset of VUE in these two cases. Cases 1,3, and 4 had evidence of chronic and recurrent asphyxia (intrathoracic effusions, cardiac dilatation, and neuronal necrosis). Case 2 had disproportionate growth restriction compared to the extent of villitis, and bone changes suggestive of an osteochondrodyplasia although radiographs were not diagnostic. The findings seemed more likely genetic than immunologic in origin, as there were no lymphocytic infiltrations.
Table 1. Summary of Clinical, Autopsy and Histopathological Findings in Four Stillbirth Cases
| Case # | Maternal Age and Parity | Maternal History | Gender and G.A of Stillborn | Autopsy Gross Findings | Microscopic Findings | Placental Findings |
| 1 | 48-year-old G4P2A1 | Chronic hypertension, gestational diabetes, morbid obesity | M, 34 weeks (twin) | Weight: 1,640 g, no malformations, small pleural, pericardial and scrotal effusions. | Chronic thymic involution | Weight: 450 g.
Fused dichorionic placenta Lymphohistiocytic villitis (involving almost all villi) with diffuse increase in perivillous fibrinoid and intervillositis. |
| 2 | 20-year-old
G1 |
Rh negative. Received Rhogam prophylaxis. | F, 30 weeks | Weight: 540 g. (SGA)
Intrauterine growth retardation syndrome1 with features of the oligohydramnios sequence (mild Potter facies, limb deformation and borderline pulmonary hypoplasia). Meconium plugging in mid small bowel. Small thymus and spleen. |
Bridging of bone trabeculae and loss of ordered columns of ballooned chondrocytes. | Weight: 120 grams. Single infarction (3 x 2 cm). Approximately 25 % of villi showing extensive lymphohistiocytic villitis and avascular villi. Advanced villous maturation.
Short umbilical cord: 25 cm. |
| 3 | 29-year-old G2P1 | Protein S deficiency treated with heparin during pregnancy | M, 36 weeks | Weight: 2,180 g. (SGA) Moderate bilateral pleural effusions and dilated cardiac chambers. Small thymus, liver and adrenals. | Neuronal necrosis in the pons and medulla. | Weight: 170 g. The majority of villi demonstrated lymphohistiocytic villitis. |
| 4 | 19-year-old G1 | Urinary tract infection in second trimester. | M, 38 weeks and 3 days | Weight: 2,140 g. Small bilateral pleural and pericardial effusions and biventricular dilation of the heart. | Growth arrest lines in the ribs. Extensive neuronal necrosis. | Weight: 270 g. Severe non-specific chronic villitis extending into stem villi, involving approximately 50% of villi. |
G.A.: Gestational Age; G= Gravidity, P= parity, A = spontaneous fetal loss; g.: grams; M: Male; F: Female; SGA: Small for gestational age. 1Diagnosis rendered by the International Skeletal Dysplasia Registry at Cedar Sinai Medical Center in Los Angeles upon evaluation of postmortem radiographs.
Are there other risks to the infant?
At least one study found an association of villitis in preterm pregnancies with PIH (29% in those <26 wks)29. However, the authors comment that villitis is uncommon in this gestation in general, a comment that requires confirmation.
Redline and Abramosky looked at 10 mothers who had recurrent villitis in a group of 59 with villitis30. They confirmed an association of villitis with IUGR, however, many of their other associations of recurrent compared to non=recurrent villitis patients were significant and provocative at P 0.5, such as increased perinatal mortality, increased maternal infection (4 with gonorrhea), obesity and autoimmune disease. The numbers of cases were very small. They found that 6 patients with basal villitis and decidual plasma cells had STD, and were unmarried and had low economic status. Another four placentas showed more intervillositis and fibrinoid and the patients had higher socio economic status, obesity, and spontaneous abortions. These findings needed collaboration from a larger study.
Perhaps the largest population confirming the association of pregnancy induced hypertension syndromes not only confirmed the association, and added that the association may be even stronger for basal villitis31. From 19,683 term placentas, 2,689 had pregnancy induced hypertension and 497 had villitis and 161 had basal villitis. Compared to 2408 with villitis and 640 basal villitis diagnoses in the remaining 16,994 placentas, the results were statistically very significant. This was a retrospective study, slides were not reviewed, and the controls were presumably all from abnormal pregnancies whose placentas were submitted to pathology for examination. The findings are only an association, and large numbers of infants with placental villitis do not have pregnancy induced hypertensive syndromes. There is much still to be learned about the complex immunology of implantation and pregnancy.
One potential but unproven risk is that moderate or severe lymphohistiocytic villitis would allow maternal T-lymphocytes to cause immune mediated disease in the fetus 3,6,32. The basis of the hypothesis was that the maternal T- lymphocytes sensitized against fetal-paternal antigens in the villi would become chimeric in the child’s blood and cause an allo-immune response that was tissue destructive and would appear to be autoimmune. Evidence for a micro-chimeric mechanism of this type has been presented for juvenile dermatomyositis and biliary atresia 33-35. My colleagues and I investigated this possibility by clinical follow up by phone at age 5 years of 14 children who has severe VUE specifically asking about potentially autoimmune symptoms such as eczema or gastrointestinal disease. The children did not differ from controls, who were similarly followed up. Our study would have been too small to have found a correlation with dermatomyositis or even if we could have followed the children into adolescence, too small to have found a correlation with systemic lupus erythematosis. There is evidence that maternal lymphocytes are often present in offspring. There is also indirect evidence that there are intrinsic protections from those lymphocytes. In a prolonged culture study of early fetal hepatic lymphocytes obtained before thymic development, there were T cells that were cytotoxic to maternal lymphocytes36. In a study of umbilical cord blood lymphocytes, fetal lymphocytes were specifically inhibitory of maternal and not just any mixed culture of lymphocytes37. In VUE, the vaso-destruction may inhibit transfer of active lymphocytes to the fetus.
What is the risk of recurrence?
The recurrence of non-symptomatic villitis in placentas is unknown. In cases where a complication has reoccurred, then the recurrence of villitis has been noted. For example, in a sequential study of 10 mothers with recurrent small for gestation infants, five had VUE in both pregnancies (although one was chronic massive intervillositis)38.
Redline reports an unusual case of recurrent villitis with positive bacterial rods on silver stain39. In the discussion he reports on 2 cases in his institution in which recurrent infection from an extra-uterine source, an entero-leiomyomatous fistula, a enteric utero fistula, and a tubo-ovarian abscess. Russell describes a single case report of mother with multiple growth retarded and fetal loss infants with last 3 documented as having VUE, although describes microsbcesses, and in one pregnancy had good result with heavy antibiotic therapy27
What is chronic intervillositis?
Chronic massive intervillositis is a pathological lesion of the placenta characterized by massive numbers of monocytes in the intervillous space that has been associated with fetal death, intrauterine growth retardation, and recurrence in subsequent pregnancies (Fig 48) 40-45.

Fig 48) The intervillous space is packed with mononuclear cells with a rim of cytoplasm (H&E)
The first time I encountered the lesion in the placenta of an extremely growth restricted stillborn infant, I thought the mother must have had a blood dyscracia. However, her peripheral blood was normal. I could not find a similar published case. A few years later at a meeting of the Society for Pediatric Pathology, Dr. Eugene Perrin presented a very similar case that had puzzled him. Others in the audience, including me, had also seen and been puzzled by similar cases. I recall after that meeting Dr. Valderrama collected cases and published the first series46.
Smaller numbers of intervillous monocytes may be observed in the placenta as an incidental finding and are of unknown clinical significance (Fig 49,50).

Fig 49) The arrow points to a focal area of intervillous monocytes in this term, AGA uncomplicated pregancy (H&E,2x)
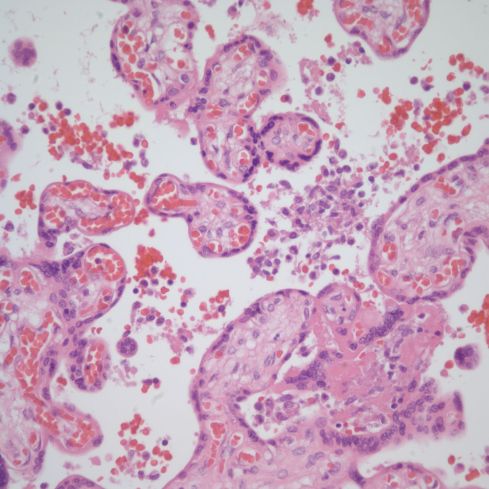
Fig 50) At higher magnification in the same case as Fig 49, a cluster of intervillous monocytes can be seen.
A variable intensity of intervillous monocytes are also present in VUE9. A study of C4d staining of placentas with villitis noted the intense staining of the syncytiotrophoblast in a placenta with massive chronic intervillositis47. Immunostaing for C4d complement antigen is the routine approach to demonstrating complement fixation in renal biopsies. Since the placenta can express paternal alloantigens on the syncytiotrophoblast surface, although not allogeneic histocompatibility antigens, a reasonable hypothesis is that massive chronic intervillositis is evidence of complement activation due to antibodies to paternal antigens on the syncytiotrophoblast microvillous surface48,49. The persistence of anti-paternal antibodies would explain the high rate of recurrence. Direct damage to the microvillous transport surface of the syncytiotrophoblast would explain the extreme growth restriction resulting from decreased nutrient transport to the fetus. Independent evidence suggests that complement activation produces cytokines that result in increased monocyte adherence to syncytiotrophoblast50.
My colleagues and I explored this potential insight of antibody induced complement fixation further by staining placentas with massive intervillositis demonstrating the key features of severe intrauterine growth restriction, fetal death and recurrence, and also placentas with asymptomatic less intense collections of intervillous monocytes51. We found that dense intervillous monocytes were associated with C4d staining in massive chronic intervillositis, and sometimes with VUE (Fig 51,52).

Fig 51) An area of massive chronic intervillositis (H&E)

FIg 52) From the same case as Fig 51, C4d staining is present over the adjacent syncytiotrophoblast border. (C4d immunostain)
Smaller numbers of monocytes seldom were associated with complement staining. The localization of C4d immunostaining to the microvillous surface of the syncytiotrophoblast suggests, as hypothesized, that a common potential allo-antigen such as transferrin on that surface might be a target. (Transferrin was proposed by Dr. Faulk’s group as the target of lymphocytes in secondary (they had a normal first pregnancy) recurrent abortion patients.) However, more difficult to explain was that C4d staining was sometimes focal within the placenta, while a surface alloantigen would be expected to be uniform. This focal inflammation might be explained if there are mechanisms that damp down the intervillous inflammation. Such a mechanism would also explain the occasional incidental clusters of intervillous macrophages.
Massive Chronic Intervillositis as a primary diagnosis based on as the name implies intense monocytic intervillositis in the context of severe growth restriction and fetal death is a rare entity. Our study suggests that C4d immuno-staining is likley to be positive. The pathologic diagnosis in the placenta is important because of its high incidence of recurrence, and the potential benefit of therapy. In our one treated case, there were milder histologic features and a good clinical outcome compared to the mother’s previous fetal losses with this lesion. The best treatment is still controversial. We suggested in our paper that both the diagnosis and monitoring of treatment might be possible with serum complement levels.
How does villitis of unknown etiology differ from infectious villitis?
Infectious villitis will be covered in a separate section. For the pathologist even without a suggestive history the finding of plasma cells in the villi is very suggestive of infection as these cells suggest processing of antigen and antibody production in situ. However, a rare plasma cell may be present in VUE. Granulomatous inflammation is common in VUE and even when it is the predominant pattern, is unlikely to be infectious in my experience (Fig 53).
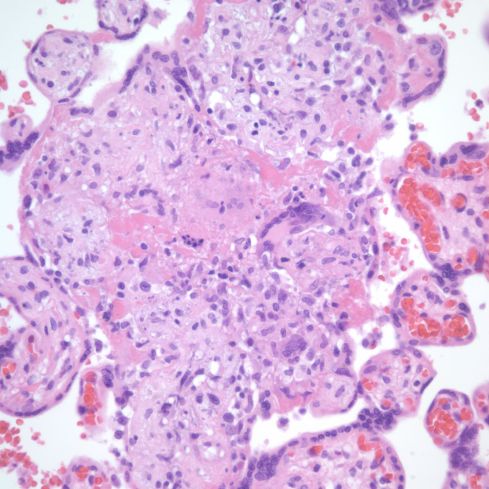
Fig 53) There is a partial thrombus in a main vessel of a small stem villus. The inflammation from VUE appears to be continuous with villi branching directly from it in the plane of section, and surrounded by other villi with VUE. (H&E 20x)
However, if there is clinical suspicion; if the mother is immuno-compromised, or if the pregnancy is from an area with endemic granulomatous disease special stains for microorganisms such as a acid fast stain or silver stain for fungi may be indicated. On H&E slides finding specific tissue features such as CMV inclusions of course overrides consideration of VUE even if it is also present. Finally, it is important for the pathologist to understand that the features of VUE may be the only finding in fetal infection (Fig 54).
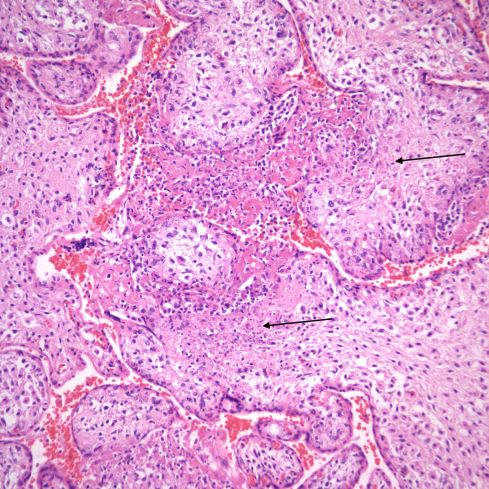
Fig 54) This cluster of villi appears to be VUE, except perhaps for some very subtle differences such as few cells with larger, irregular nuclei, and some areas of coagulation necrosis in the villi. I don’t think prospectively that I would have recognized this for a villitis associated with toxoplasmosis infection of the infant, which it was. (H&E, 20x)
References:
- Altshuler G, Russell P. The human placental villitides: A review of chronic intrauterine infection. Curr Top Pathol 1975;60:63-112.
- Mortimer G, MacDonald DJ, Smeeth A. A pilot study of the frequency and significance of placental villitis. Br J Obstet Gynaecol 1985;92:629-33.
- Altemani AM. Immunohistochemical study of the inflammatory infiltrate in villitis of unknown etiology. A qualitative and quantitative analysis. Pathol Res Pract 1992;188:303-9.
- Labarrere CA, McIntyre JA, Faulk WP. Immunohistologic evidence that villitis in human normal term placentas is an immunologic lesion. Am J Obstet Gynecol 1990;162:515-22.
- Katzman PJ, Murphy SP, Oble DA. Immunohistochemical analysis reveals an influx of regulatory T cells and focal trophoblastic STAT-1 phosphorylation in chronic villitis of unknown etiology. Pediatr Dev Pathol 2011;14:284-93.
- Redline RW, Patterson P. Villitis of unknown etiology is associated with major infiltration of fetal tissue by maternal inflammatory cells [see comments]. Am J Pathol 1993;143:473-9.
- Myerson D, Parkin RK, Benirschke K, Tschetter CN, Hyde SR. The pathogenesis of villitis of unknown etiology: analysis with a new conjoint immunohistochemistry-in situ hybridization procedure to identify specific maternal and fetal cells. Pediatr Dev Pathol 2006;9:257-65.
- Labarrere CA, Faulk WP. Maternal cells in chorionic villi from placentae of normal and abnormal human pregnancies. Am J Reprod Immunol 1995;33:54-9.
- Russell P. Inflammatory lesions of the human placenta. III. The histopathology of villitis of unknown aetiology. Placenta 1980;1:227-44.
- Jacques S, Qureshi F. Chronic villitis of unknown etiology in twin gestations. Pediatr Pathol 1994;14:575-84.
- De Tar MW, Klohe E, Grosset A, Rau T. Neonatal alloimmune thrombocytopenia with HLA alloimmunization: case report with immunohematologic and placental findings. Pediatr Dev Pathol 2002;5:200-5.
- Beer AE, Billingham RE. Maternally acquired runt disease. Science 1973;179:240-3.
- Beer AE, Billingham RE, Yang SL. Maternally induced transplantation immunity, tolerance, and runt disease in rats. J Exp Med 1972;135:808-26.
- Billingham RE, Brown JB, Defendi V, Silvers WK, Steinmuller D. Quantitative studies on the induction of tolerance of homologous tissues and on runt disease in the rat. Ann N Y Acad Sci 1960;87:457-71.
- Labarrere C, Faulk W. Anchoring Villi in Human Placental Basal Plate – Lymphocytes; Macrophages and Coagulation. Placenta12 1991:173-82.
- Knox WF, Fox H. Villitis of unknown aetiology: its incidence and significance in placentae from a British population. Placenta 1984;5:395-402.
- Redline RW. Villitis of unknown etiology: noninfectious chronic villitis in the placenta. Hum Pathol 2007;38:1439-46.
- Labarrere CA, Faulk WP, McIntyre JA. Villitis in normal term human placentae: frequency of the lesion determined by monoclonal antibody to HLA-DR antigen. J Reprod Immunol 1989;16:127-35.
- Althalbe O, Labarrere C. Chronic villitis of unknown aetiology and intrauterine growth-retarded infants of normal and low ponderal index. Placenta 1985;6:369-73.
- Nordenvall M, Sandstedt B. Placental villitis and intrauterine growth retardation in a Swedish population. Apmis 1990;98:19-24.
- Bjoro K, Jr., Myhre E. The role of chronic non-specific inflammatory lesions of the placenta in intra-uterine growth retardation. Acta Pathol Microbiol Immunol Scand [A] 1984;92:133-7.
- Becroft DM, Thompson JM, Mitchell EA. Placental villitis of unknown origin: epidemiologic associations. Am J Obstet Gynecol 2005;192:264-71.
- Derricott H, Jones RL, Heazell AE. Investigating the association of villitis of unknown etiology with stillbirth and fetal growth restriction – a systematic review. Placenta 2013;34:856-62.
- Korteweg FJ, Erwich JJ, Holm JP, et al. Diverse placental pathologies as the main causes of fetal death. Obstet Gynecol 2009;114:809-17.
- Rayburn W, Sander C, Jr MB, Rygiel R. The stillborn fetus: Placental histologic examination in determining a cause. Obstet Gynecol 1985;65:637-41.
- Gunyeli I, Erdemoglu E, Ceylaner S, Zergeroglu S, Mungan T. Histopathological analysis of the placental lesions in pregnancies complicated with IUGR and stillbirths in comparison with noncomplicated pregnancies. J Turk Ger Gynecol Assoc 2011;12:75-9.
- Russell P, Atkinson K, Krishnan L. Recurrent reproductive failure due to severe placental villitis of unknown etiology. J Reprod Med 1980;24:93-8.
- Bendon R, Potenziani-Pradella S. To the Editor. Pediatr Dev Pathol 2015;18:426.
- Hansen AR, Collins MH, Genest D, et al. Very low birthweight Infant’s placenta and its relation to pregnancy and fetal characteristics. Pediatr Dev Pathol 2000;3:419-30.
- Redline R, Abramowsky C. Clinical and pathologic aspects of recurrent placental villitis. Hum Pathol 1985;16:727-31.
- Katzman PJ, Blitman J, Metlay LA. Basal Chronic Villitis and Disorders of the Placental Basal Plate: A Possible Immunological Link Between Hypertensive Disorders of Pregnancy and Morbidly Adherent Placenta. Pediatr Dev Pathol 2019;22:334-9.
- Labarrere CA, McIntyre JA, Faulk WP. Immunohistologic evidence that villitis in human normal term placentas is an immunologic lesion. Am J Obstet Gynecol 1990;162:515-22.
- Reed AM, McNallan K, Wettstein P, Vehe R, Ober C. Does HLA-dependent chimerism underlie the pathogenesis of juvenile dermatomyositis? J Immunol 2004;172:5041-6.
- Reed AM, Picornell YJ, Harwood A, Kredich DW. Chimerism in children with juvenile dermatomyositis. Lancet 2000;356:2156-7.
- Muraji T, Hosaka N, Irie N, et al. Maternal microchimerism in underlying pathogenesis of biliary atresia: quantification and phenotypes of maternal cells in the liver. Pediatrics 2008;121:517-21.
- Miyagawa Y, Matsuoka T, Baba A, et al. Fetal liver T cell receptor gamma/delta+ T cells as cytotoxic T lymphocytes specific for maternal alloantigens. J Exp Med 1992;176:1-7.
- Brune T, Beier K, Exeler R, Harms E, Louwen F. Neonatal lymphocytes dominate against lymphocytes of their own mother but not against allogenic maternal or adult lymphocytes in bidirectional mixed lymphocyte cultures. Fetal Diagn Ther 2003;18:154-9.
- Labarrere C, Althabe O. Chronic villitis of unknown aetiology in recurrent intrauterine fetal growth retardation. Placenta 1987;8:167-73.
- Redline R. Recurrent villitis of bacterial etiology. Pediatr Pathol Lab Med 1996;16:995-1001.
- Labarrere C, Mullen E. Fibrinoid and trophoblastic necrosis with massive chronic intervillositis: an extreme variant of villitis of unknown etiology. Am J Reprod Immunol Microbiol 1987;15:85-91.
- Jacques SM, Qureshi F. Chronic intervillositis of the placenta. Arch Pathol Lab Med 1993;117:1032-5.
- Doss BJ, Greene MF, Hill J, Heffner LJ, Bieber FR, Genest DR. Massive chronic intervillositis associated with recurrent abortions. Hum Pathol 1995;26:1245-51.
- Redline RW, Zaragoza M, Hassold T. Prevalence of developmental and inflammatory lesions in nonmolar first-trimester spontaneous abortions. Hum Pathol 1999;30:93-100.
- Boyd TK, Redline RW. Chronic histiocytic intervillositis: a placental lesion associated with recurrent reproductive loss. Hum Pathol 2000;31:1389-96.
- Contro E, deSouza R, Bhide A. Chronic intervillositis of the placenta: a systematic review. Placenta 2010;31:1106-10.
- Valderrama E. Massive chronic intervillositis: Report of three cases. Lab Invest 1992;66:10P.
- Rudzinski E, Gilroy M, Newbill C, Morgan T. Positive C4d immunostaining of placental villous syncytiotrophoblasts supports host-versus-graft rejection in villitis of unknown etiology. Pediatr Dev Pathol 2013;16:7-13.
- Szekeres-Bartho J. Immunological relationship between the mother and the fetus. Int Rev Immunol 2002;21:471-95.
- McIntyre JA, Faulk WP, Verhulst SJ, Colliver JA. Human trophoblast-lymphocyte cross-reactive (TLX) antigens define a new alloantigen system. Science 1983;222:1135-7.
- Xiao J, Garcia-Lloret M, Winkler-Lowen B, Miller R, Simpson K, Guilbert LJ. ICAM-1-mediated adhesion of peripheral blood monocytes to the maternal surface of placental syncytiotrophoblasts: implications for placental villitis. Am J Pathol 1997;150:1845-60.
- Bendon RW, Coventry S, Thompson M, Rudzinski ER, Williams EM, Oron AP. Significance of C4d Immunostaining in Placental Chronic Intervillositis. Pediatr Dev Pathol 2015;18:362-8.
Addendum
Since posting I found a few more unscanned images that I wanted to share.

The central villus shows that the lymphocytes just beneath the syncytium are T-lymphocytes, and are associated with a breach in a the lower portion of the syncytium. (CD-3, 20x)
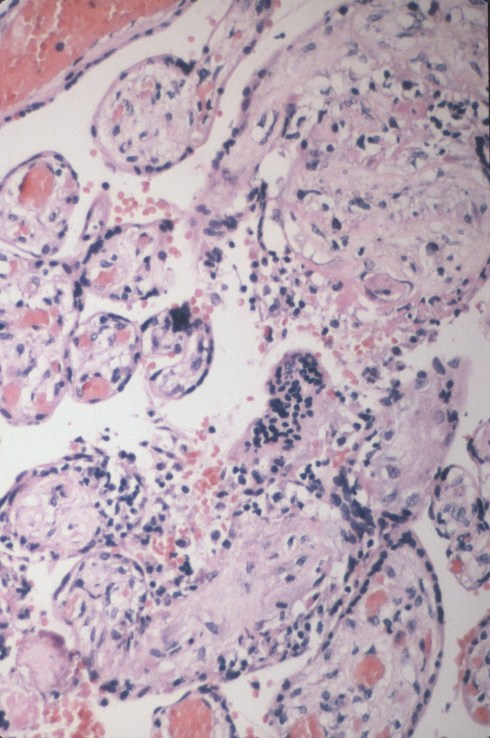
This image is from the first pregnancy of a mother whose second infant died of the complications of Rh alloimmunization (Immune Hydrops). Is VUE part of the sensitization process? Fortunately, I did not have enough cases to explore this due to adequate postpartum therapy of RH- mothers.(H&E)
