Gross Findings:
The kidneys are visible from the posterior organ block. The location, amount of fat, color, and the hilar connection of the ureters, renal artery and renal vein all can be noted. Since this manual is not concerned with fetal malformation or genetic diseases per se, they will be only briefly mentioned.
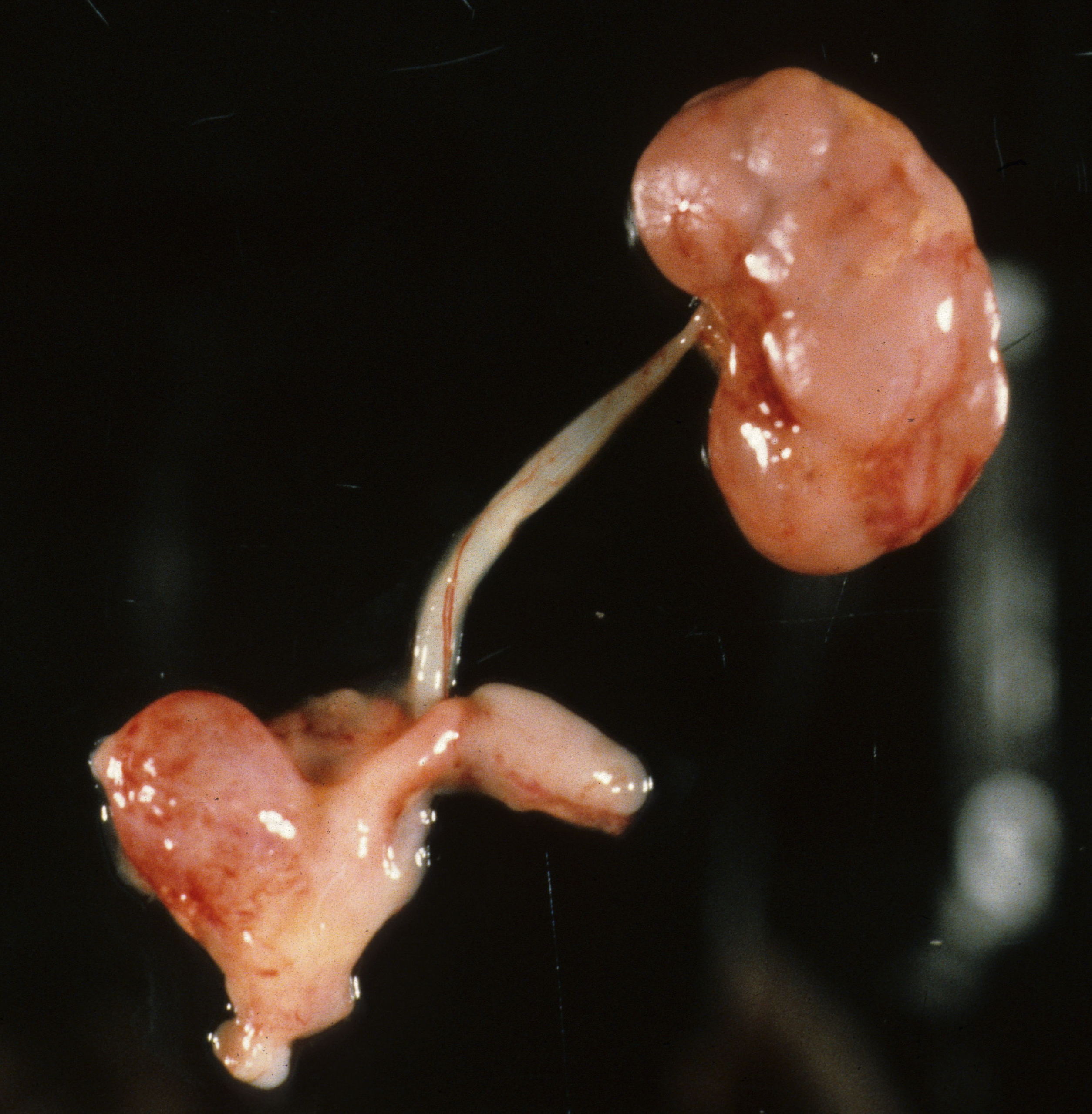
Absent kidneys: Absent kidneys occurs with multiple genetic pathways. Beside the obvious lack of the kidneys, the distortion of the overlying adrenal glands also is evidence of the absent kidneys during development. (Fig 1).
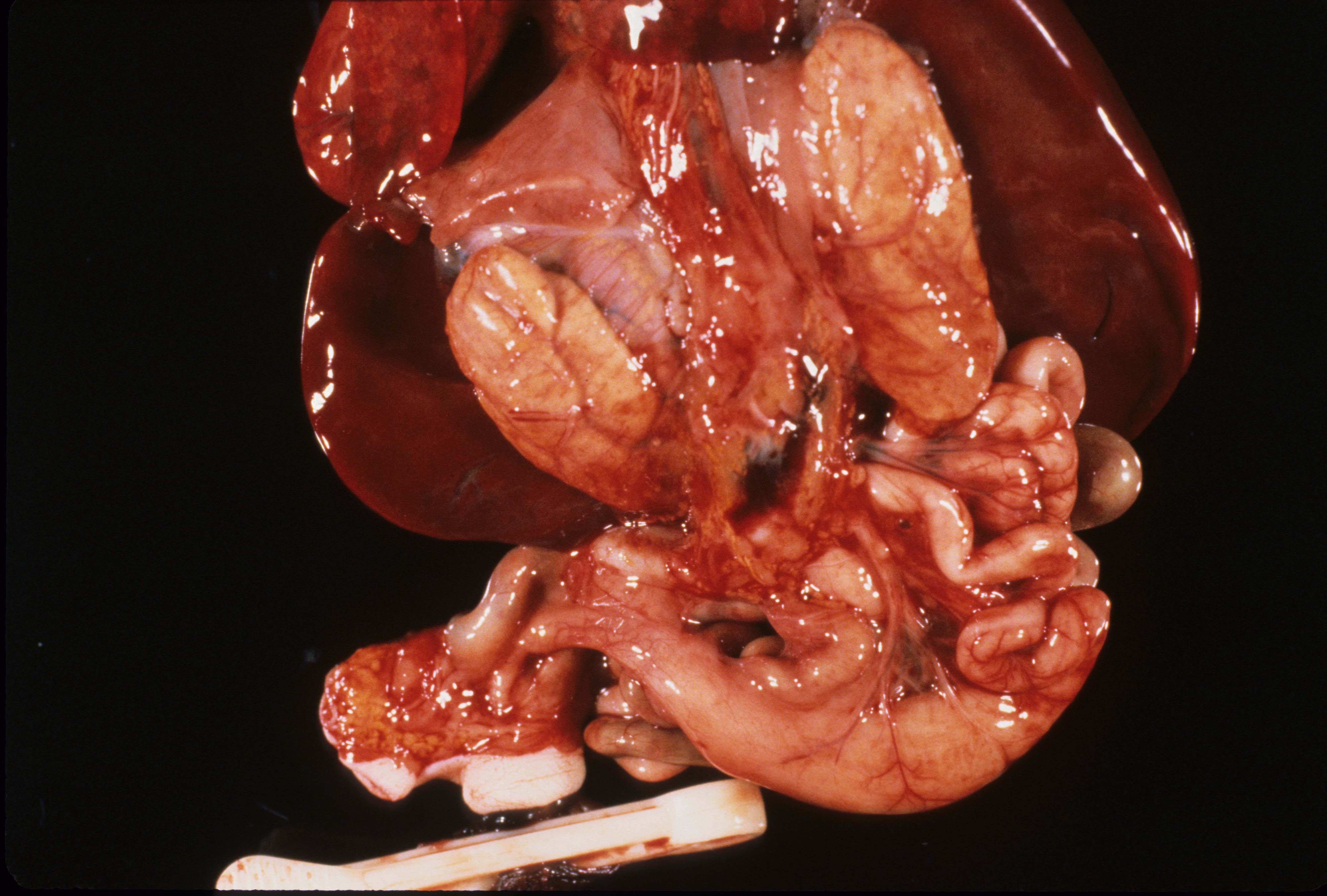
A kidney may be absent on only one side often with associated anomalies including absence of the ipsilateral side of the uterus and Fallopian tube. (Fig 2)
Fused kidneys (Horseshoe kidneys): The fusion of the kidneys usually occurs in the midline of the lower pole and they may be ectopic in that they are caudad in location. The anomaly is more common in Monosomy X (Turner syndrome). (Fig 3)

Ectopic kidneys: The position of the kidney can show abnormalities presumably from development and most commonly is near the pelvis. (Fig 4)
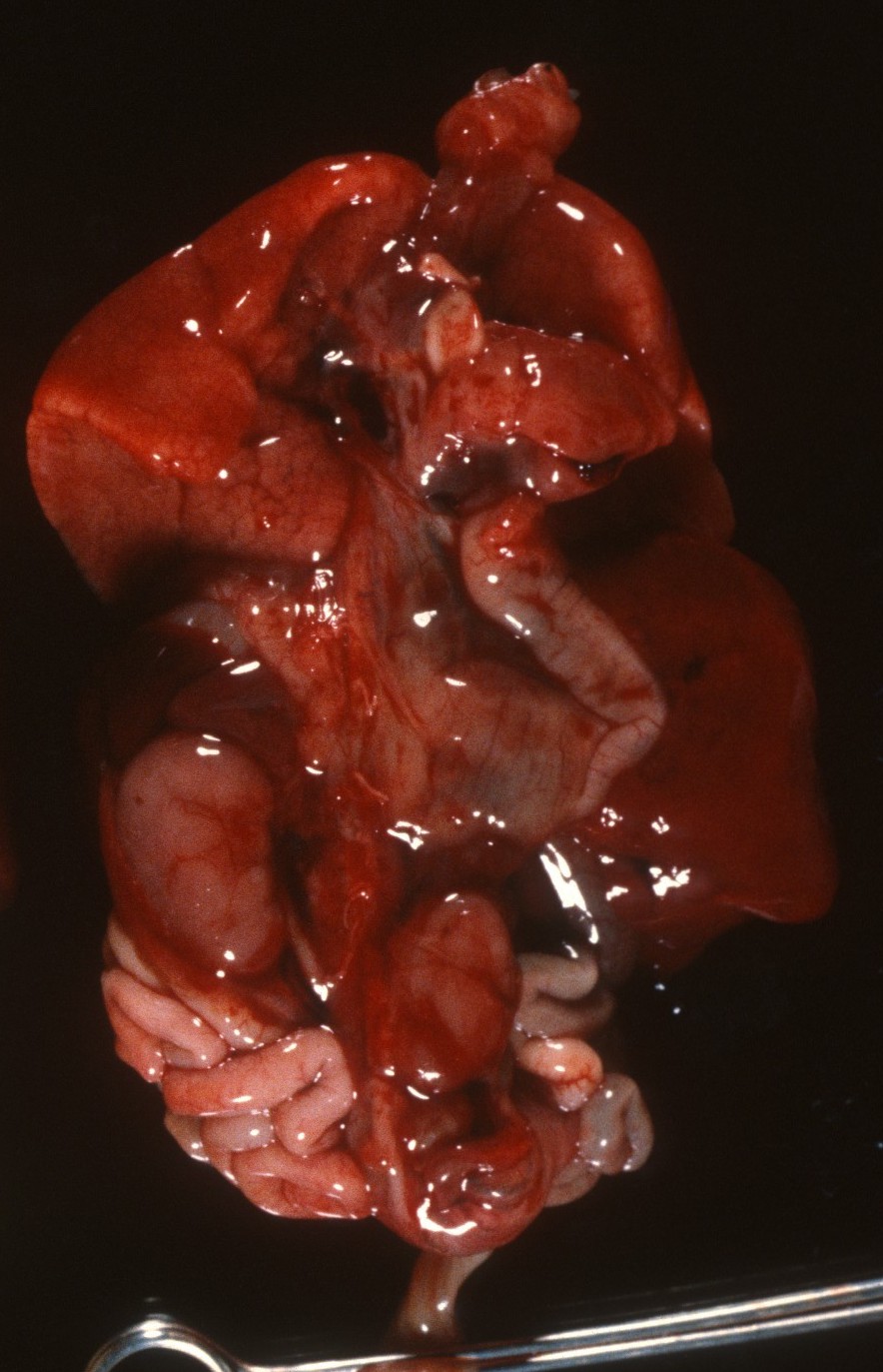
Small or enlarged kidney: As with all organs, the kidneys stripped of perirenal fat are weighed and compared to norms including the kidney to brain ratio. Massive congestion and hemorrhage can result in large kidneys as well as various form of renal cystic disease (Fig 5).

In other cases, developmental anomalies result in small kidneys. One factor that delays renal tubular development (renal tubular dysgenesis) produces loss of or decreased proximal tubule development (see supplement “A” below). If the kidneys are symmetric, they can be weighted together, but if asymmetrical, they need to be weighed separately. (Fig 6)

Increased perirenal adipose tissue: With macrosomia in the infants of diabetic mothers, there is prominent subcutaneous adipose tissue, especially in the cheeks, but also over the pericardium, and kidney. (Fig 7)
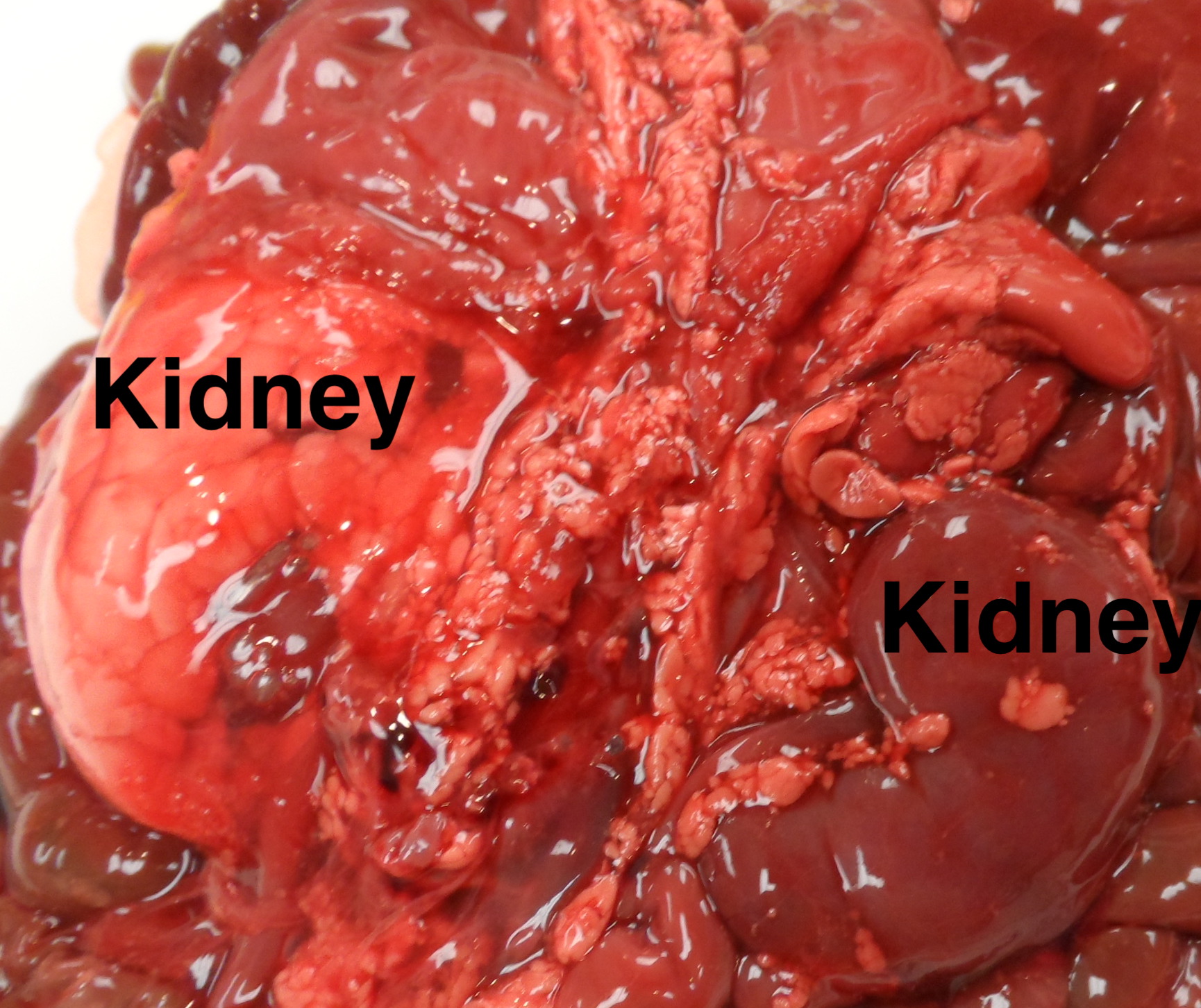
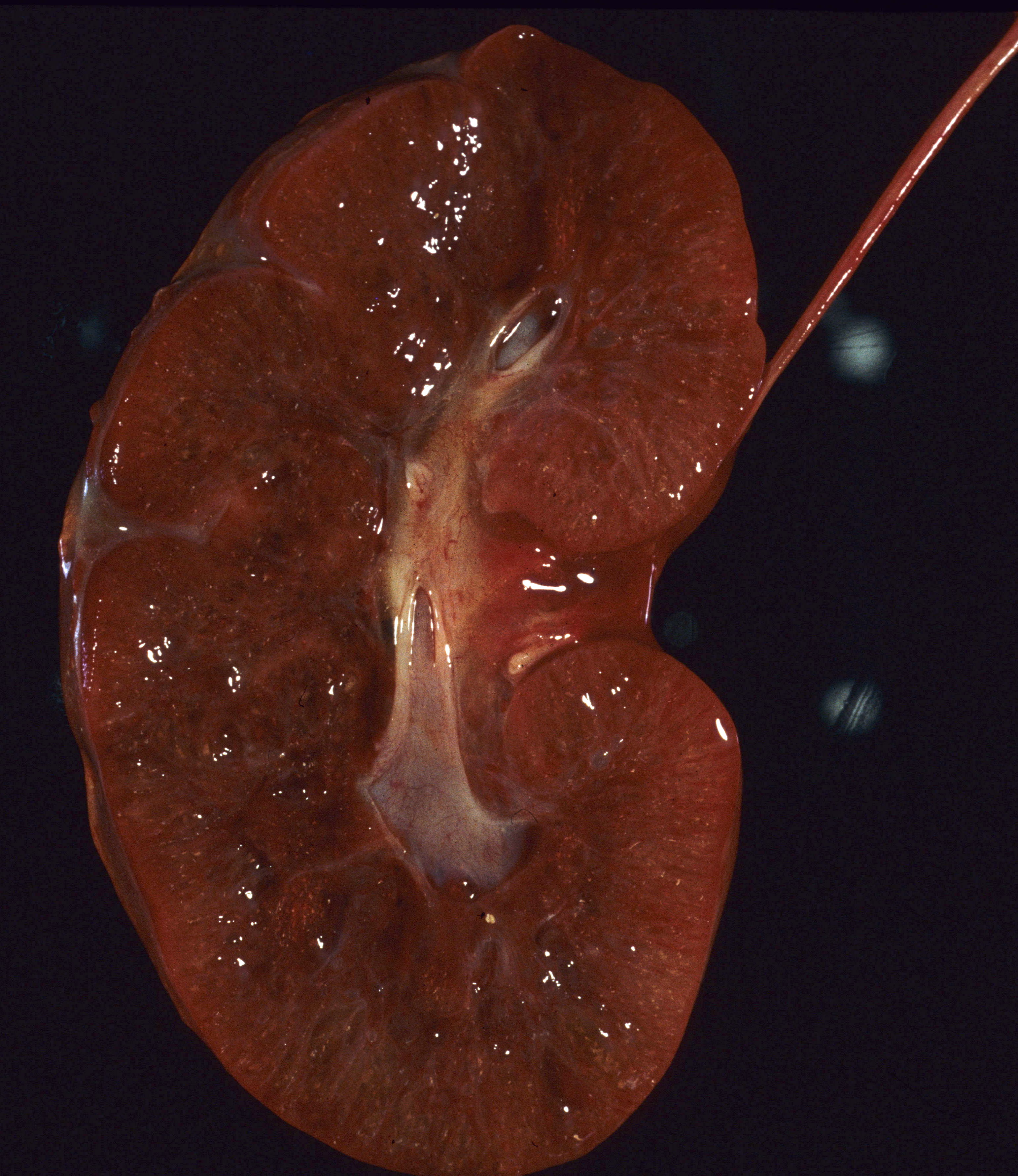
Discolored kidney: Infarctions, umbilical vein thrombus, cystic disease or massive congestion/hemorrhage (Fig 5) can alter the color of the kidney making it pale or deeper red (Fig 8).

Masses: The kidney may demonstrate Wilms tumor, congenital mesoblastic nephroma, or solitary cysts.
Dissection and sampling:
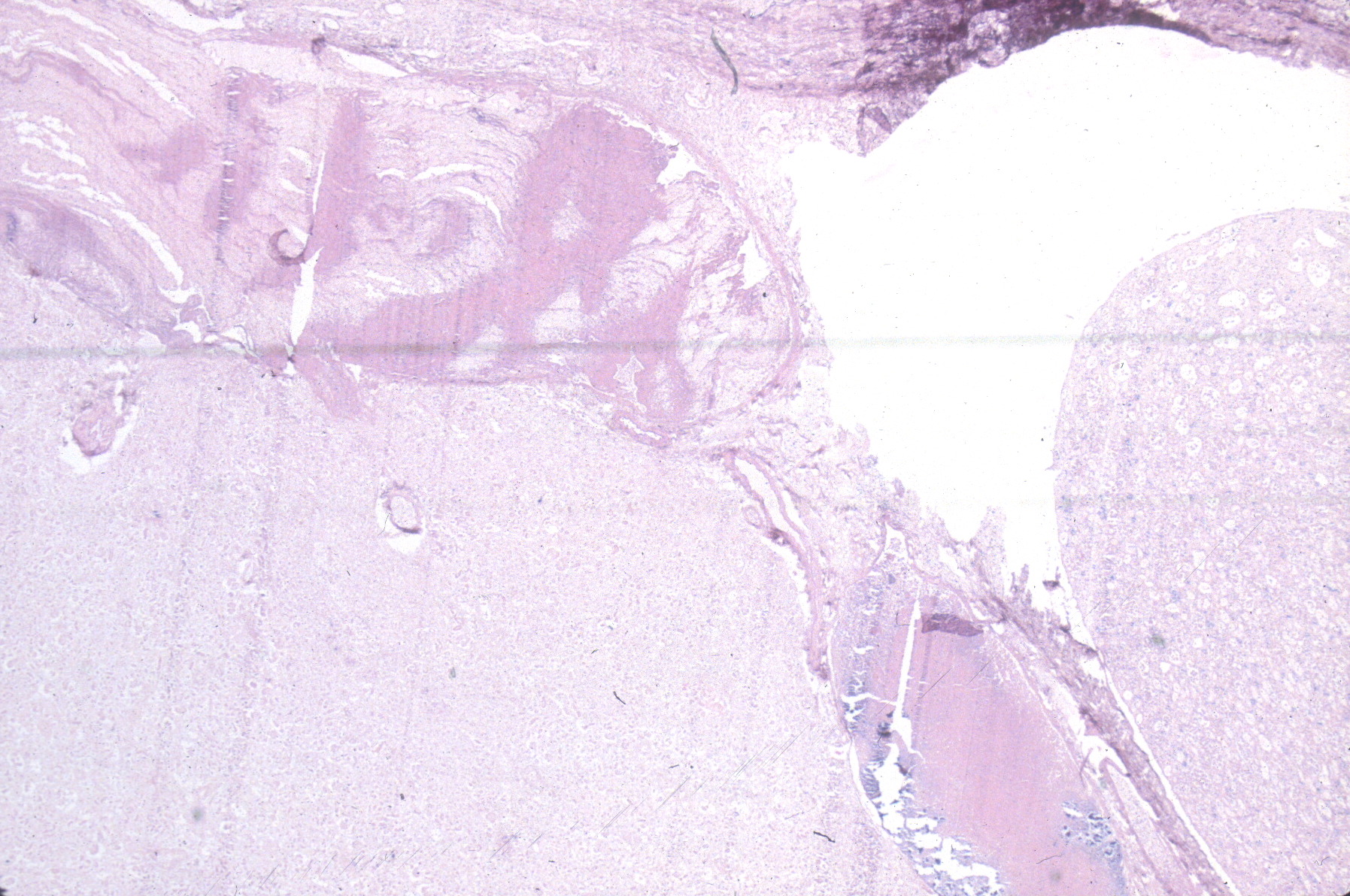
The renal vein is opened, and the ureters and renal artery are examined externally. The kidney is then removed and weighed. It can then be split longitudinally (bivalved) to reveal internal structure, particularly patterns of cystic disease and hemorrhage (Fig 9a-c).
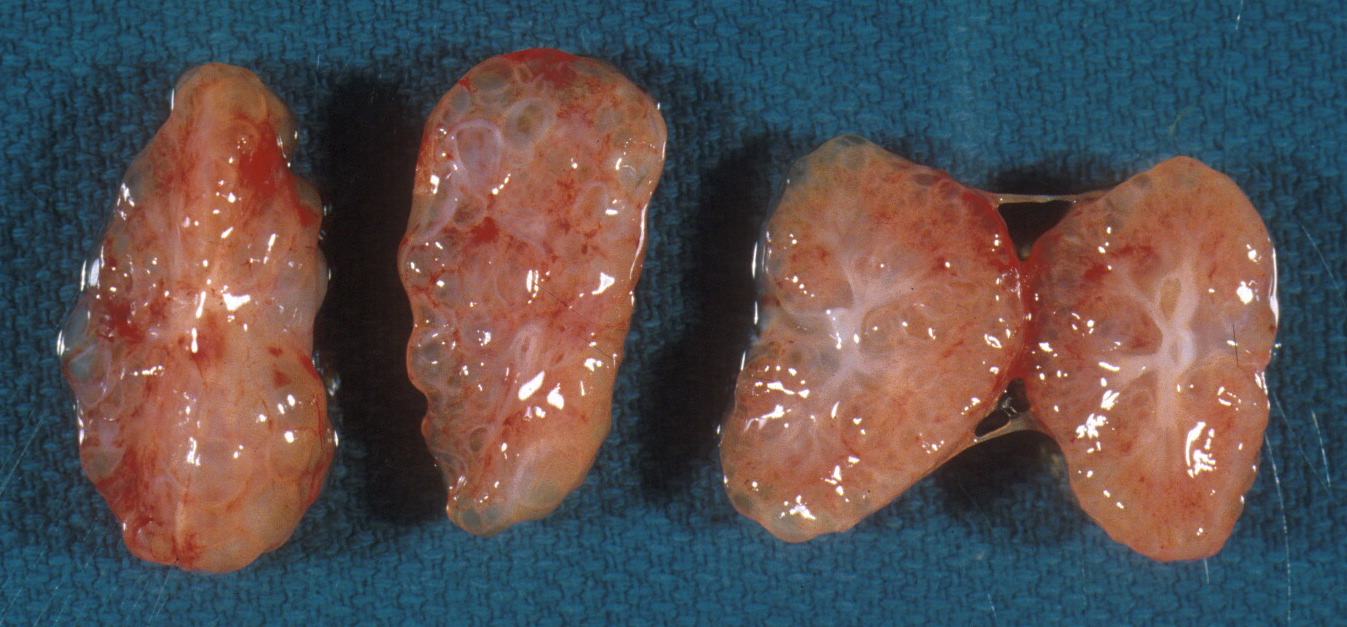

Histologic samples taken parallel to the cut surface or perpendicular to it in larger kidneys will best reveal generations of nephrons. Focal lesions can be sampled.
Oligohydramnios sequence (Potter syndrome):
Oligohydramnios is associated with an increased risk of stillbirth. A major cause of oligohydramnios is lack of urine production or obstructed urinary outflow. Intrinsic renal causes include autosomal recessive polycystic kidneys, and renal tubular dysplasia which require microscope diagnosis. While some multicystic dysplasia is intrinsic as in familial aplasia/dysplasia, often it is secondary to obstruction at some level of the urinary tract. In such cases, the entire urinary tract needs to be evaluated for the correct diagnosis. The normal diameter of the ureters appears wide for the size of the kidney, but this may be due to the need for a low resistance tube. Hydronephrosis does occur but the ureter will be clearly distended, and the calyces will also appear thickened. The insertion of the ureter into the bladder may be abnormal. In obstruction to the outflow proximal from the kidneys, the bladder will be hypoplastic, but urethral obstruction will usually demonstrate a large bladder. The latter may rupture into the abdomen or through the urachus to the umbilical cord. The bladder with possible urethral obstruction is best opened from the apex toward the urethra so that the obstruction (posterior urethral valves, atresia or outflow mass can be visualized prior to cutting through it. Another useful technique that also works on hypoplastic bladders is to inject the bladder in situ with radiocontrast and take AP and lateral films (Fig 10).

Oligohydramnios sequence can also occur with prolonged rupture of fetal membranes or chronically decreased blood flow, as in chronic twin to twin transfusion. To develop the full oligohydramnios sequence (Potter facies, limb deformities, pulmonary hypoplasia, and amnion nodosum) requires almost complete anhydramnios, not just a decreased amount of fluid. The topic of oligohydramnios will be covered more fully in the obstetrical pathology blog under lethal fetal disease. (See supplement “B” below from an older research project I had written on oligohydramnios)
Microscopic Features:
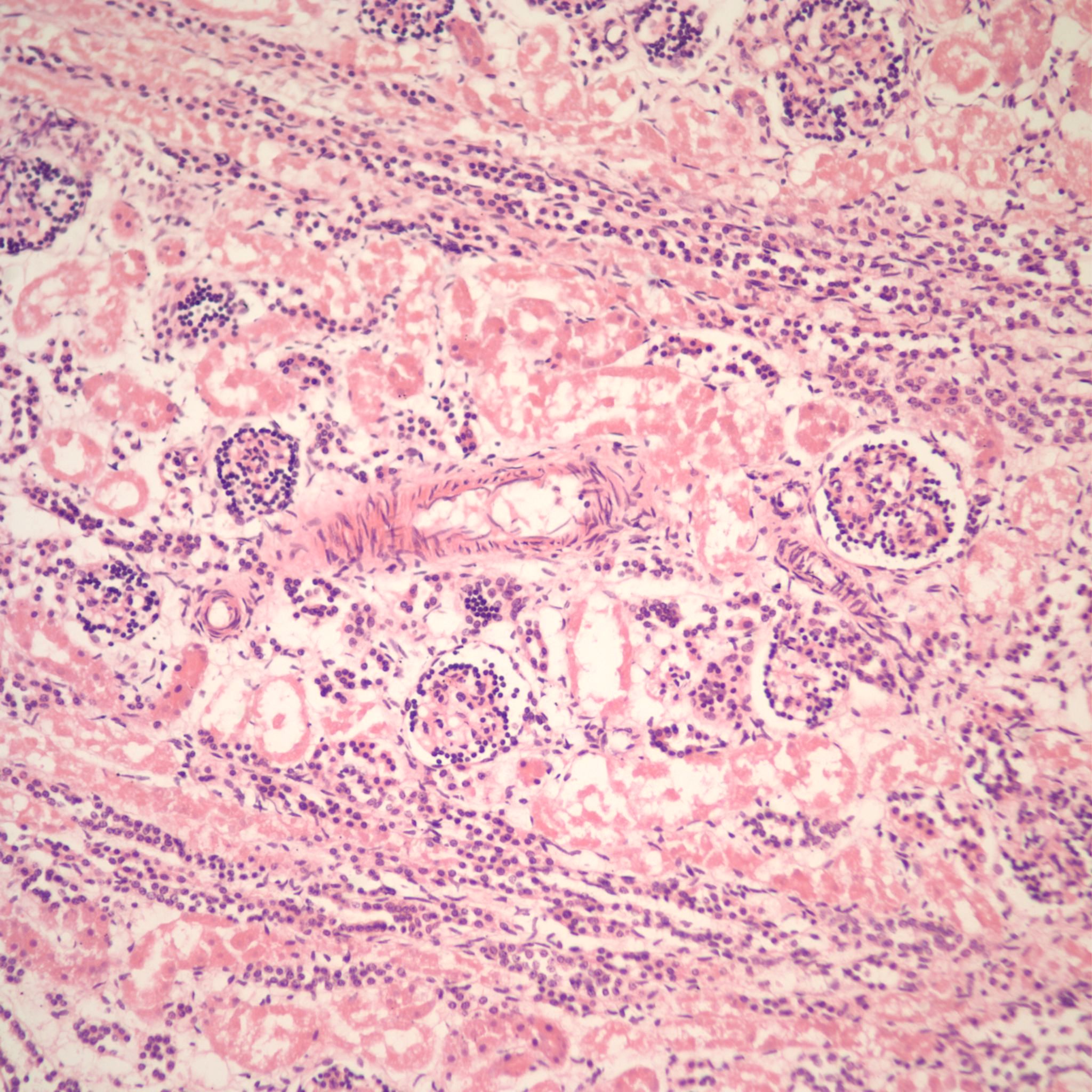
Duration of postmortem retention: The loss of 1% of tubular basophilia occurs after 4 hours of postmortem retention and usually involves the straight and convoluted proximal tubules (Fig 11).
Complete loss of renal basophilia does not occur until 4 weeks of retention (Fig 12)

Gestational age: The generations of nephrons branch in sequence from the urethral bud induction in the mesenchyme. Counting the generations had been proposed as a method of assessing gestational age. One simple extension of this is that after 35 weeks of gestation nephrogenesis ceases, and this is evidence of maturity[1, 2] (Fig 13 a,b).

Renal vein thrombosis: The renal vein appears to be particularly susceptible in utero to thrombosis. This is most common in infants of diabetic mothers, but occurs in other thrombogenic circumstances as well (Fig 14a, b).
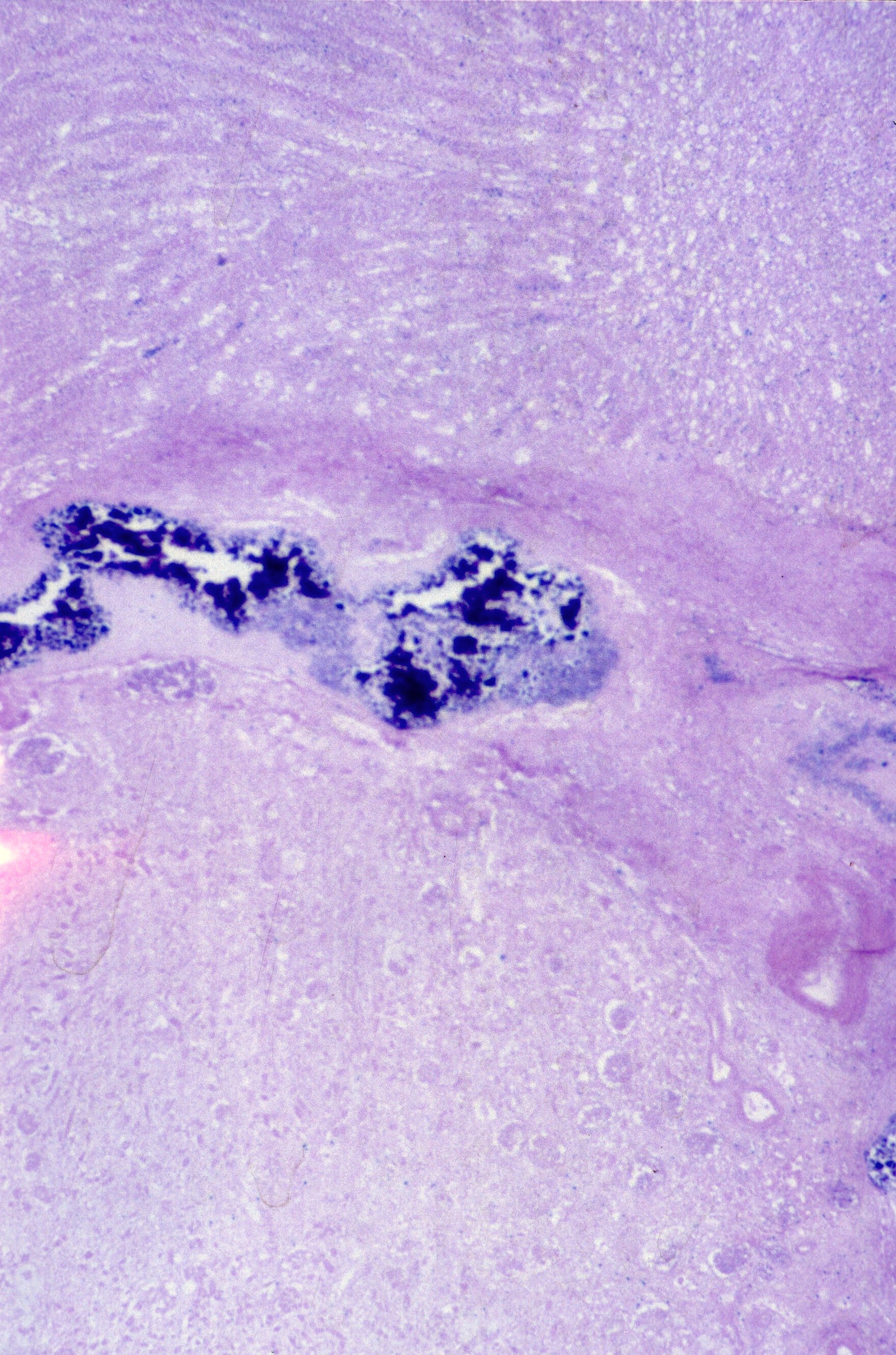
The basis for the localization of thrombus in the renal vein in infants of a diabetic mother is unclear, but if glucosuria is part of the mechanism of polyhydramnios, it is possible that the blood leaving the kidney has had serum extracted to the point of being polycythemic and hyperviscous.
Congestion and hemorrhage: The kidney can show passive congestion in heart failure as in other organs, and occasionally hemorrhage. The congestion is often confined to the cortico- medullary junction, likely due to the shunting of arterial blood away from the cortex in asphyxia (15a, b).


Hemorrhages can also occur with septic shock and presumed disseminated intravascular coagulation (Fig 15c).

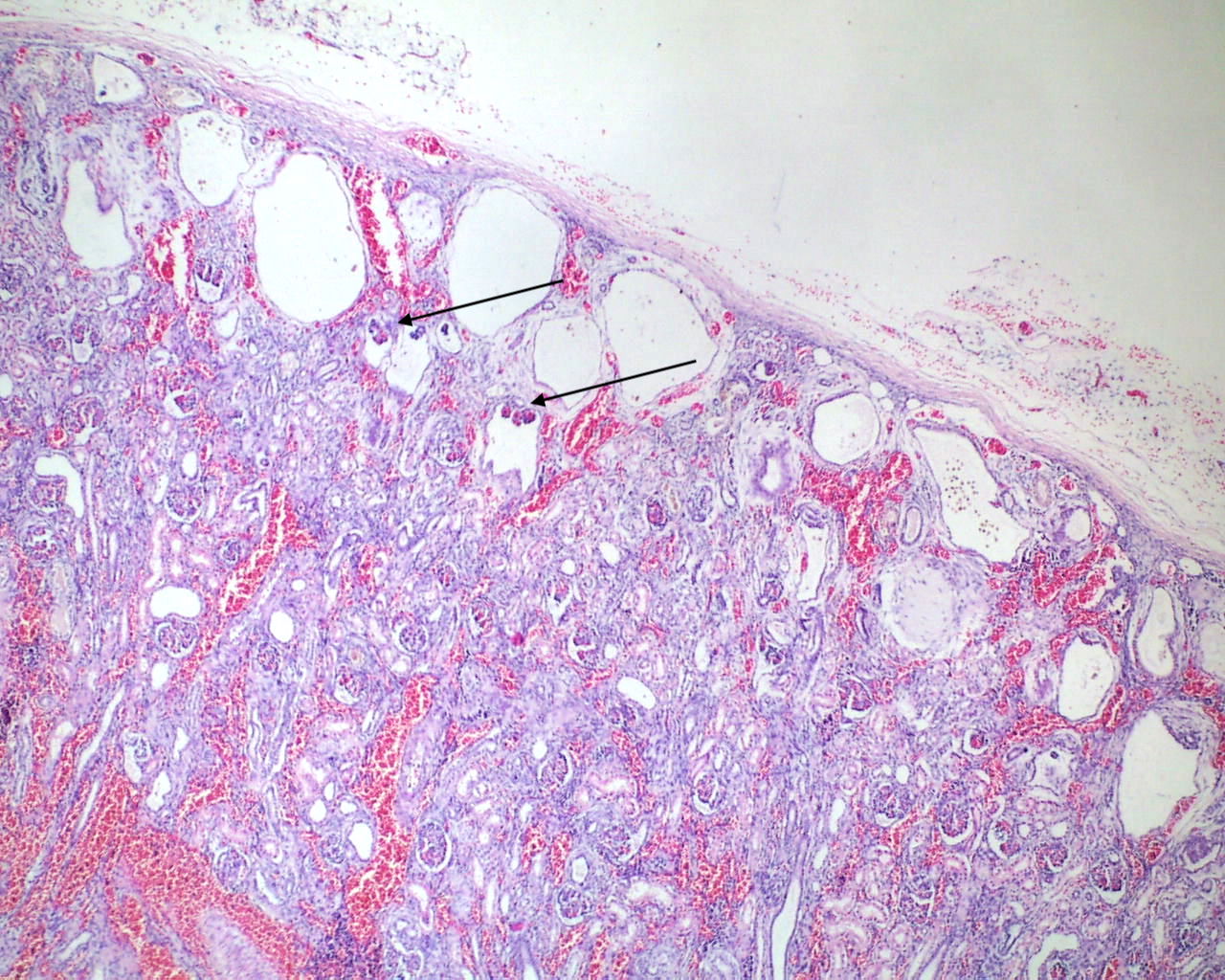
Mineralization within renal tubules: In some infants there are mineralized deposits of calcium and iron in the tubules. The most logical explanation is that these are residual hemorrhagic casts from a prior episode of acute tubular necrosis. (Fig 16a-c)
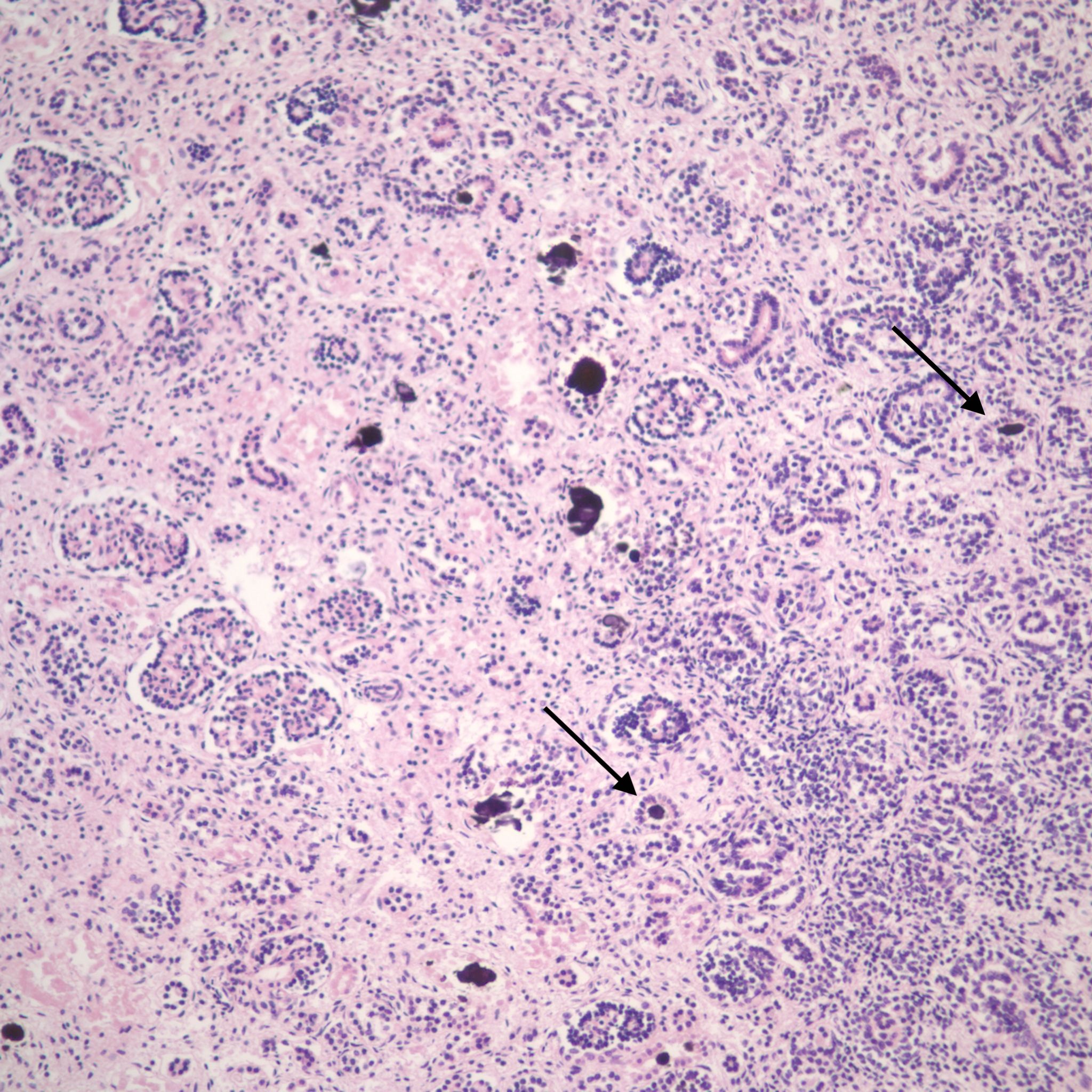
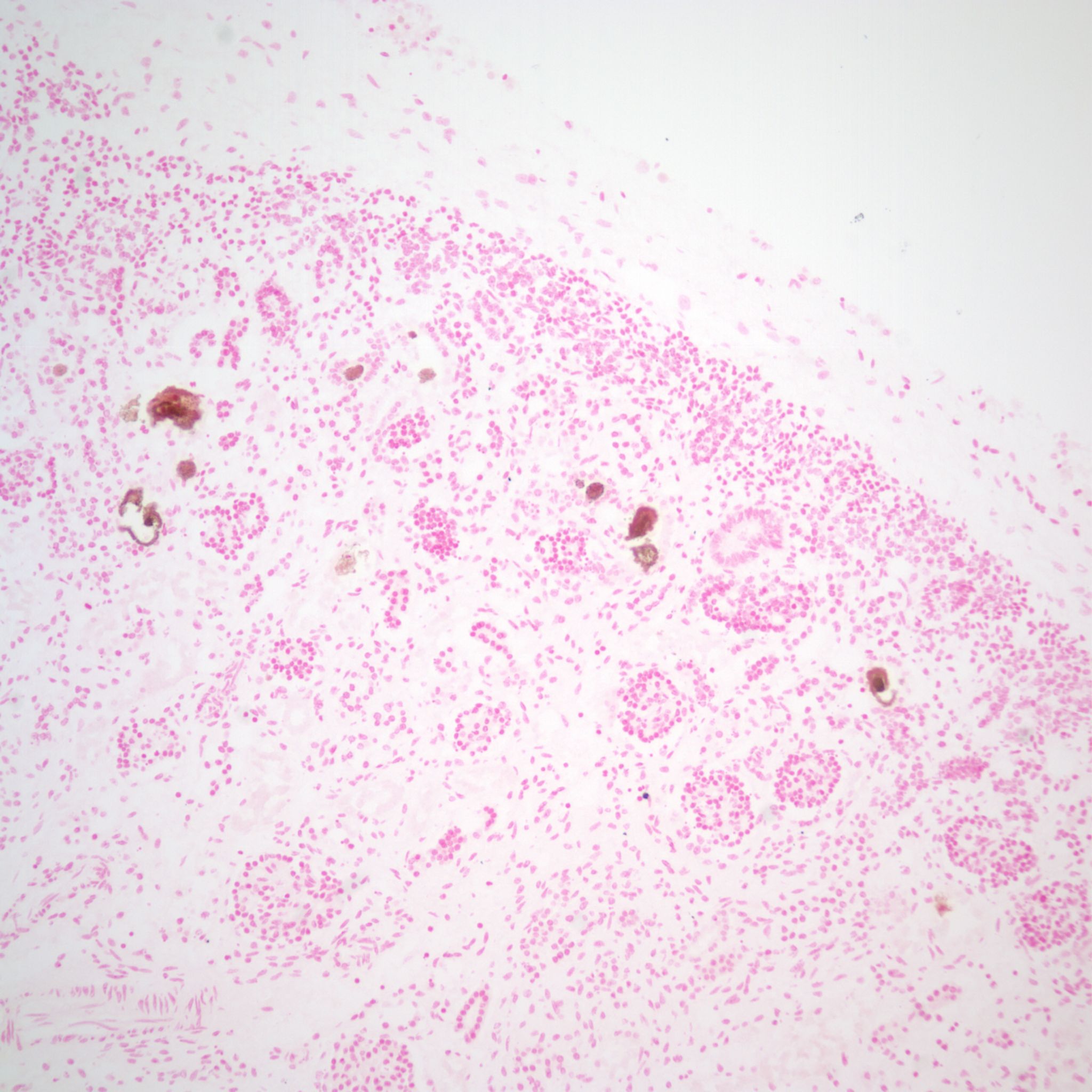
A quick survey of renal cystic disease:
Cystic Dysplasia (Fig 17)

Autosomal Recessive polycystic disease (Fig 18)
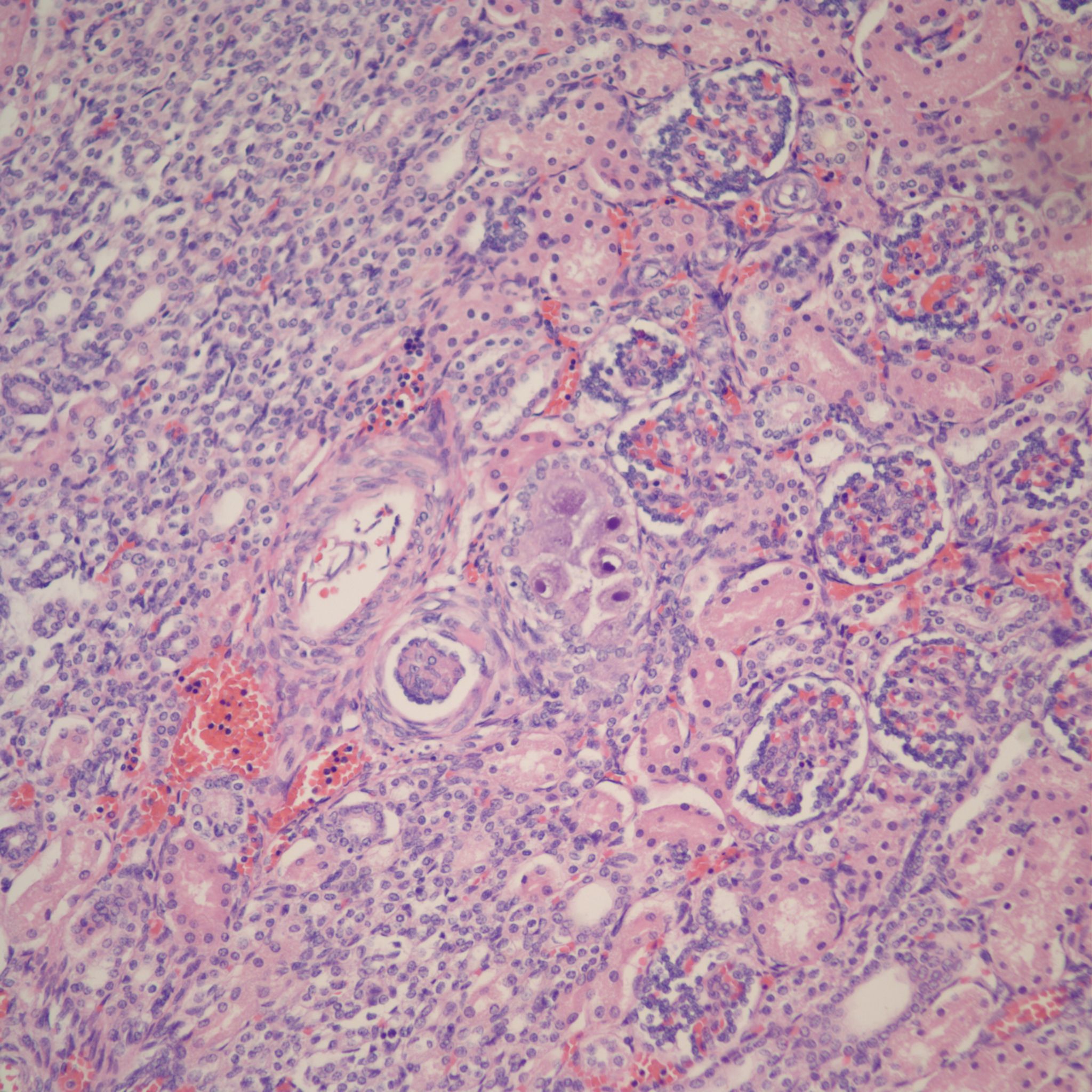
Glomerulocystic disease (Fig 19)
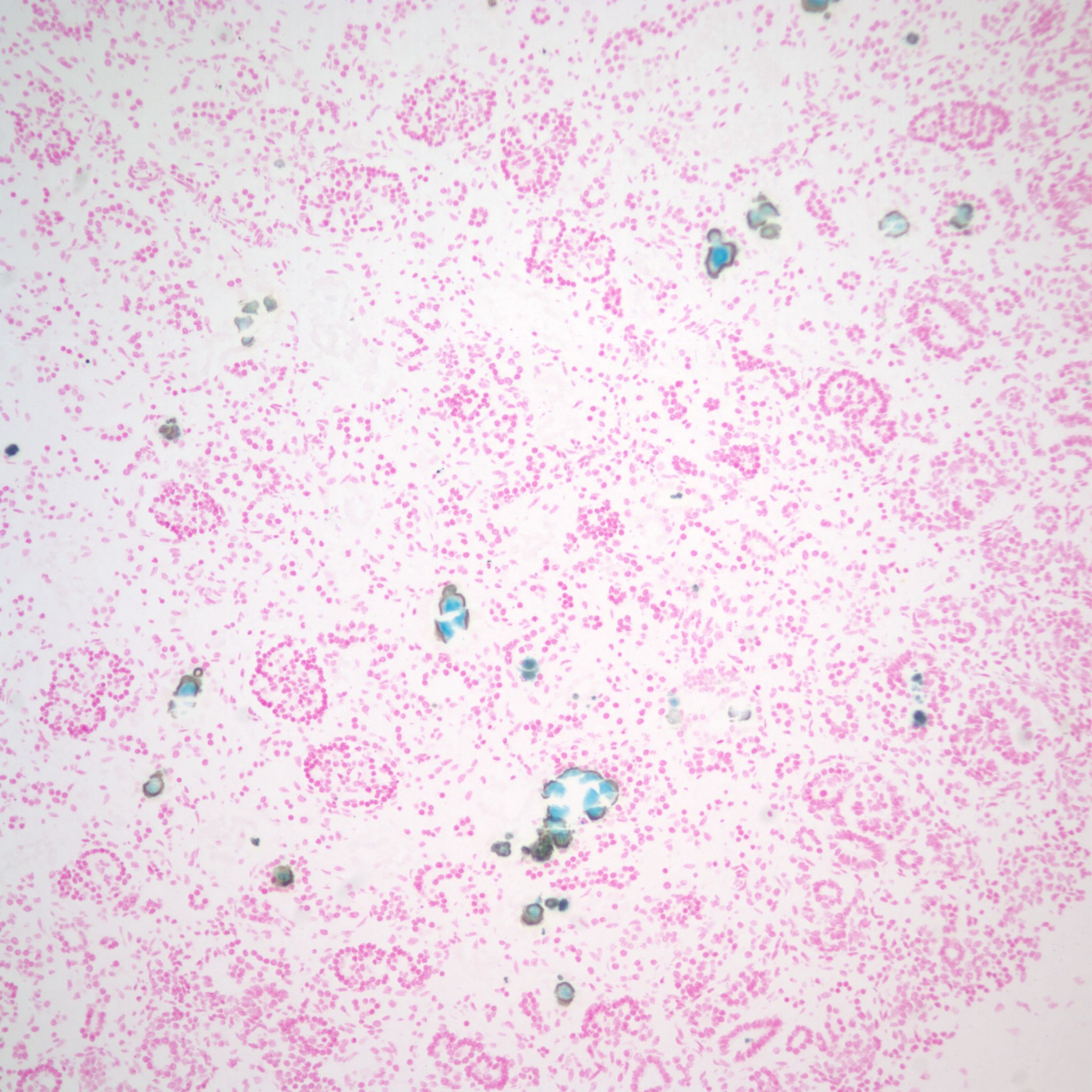
Infection: The renal tubules are a common target of congenital CMV infection (Fig 20).
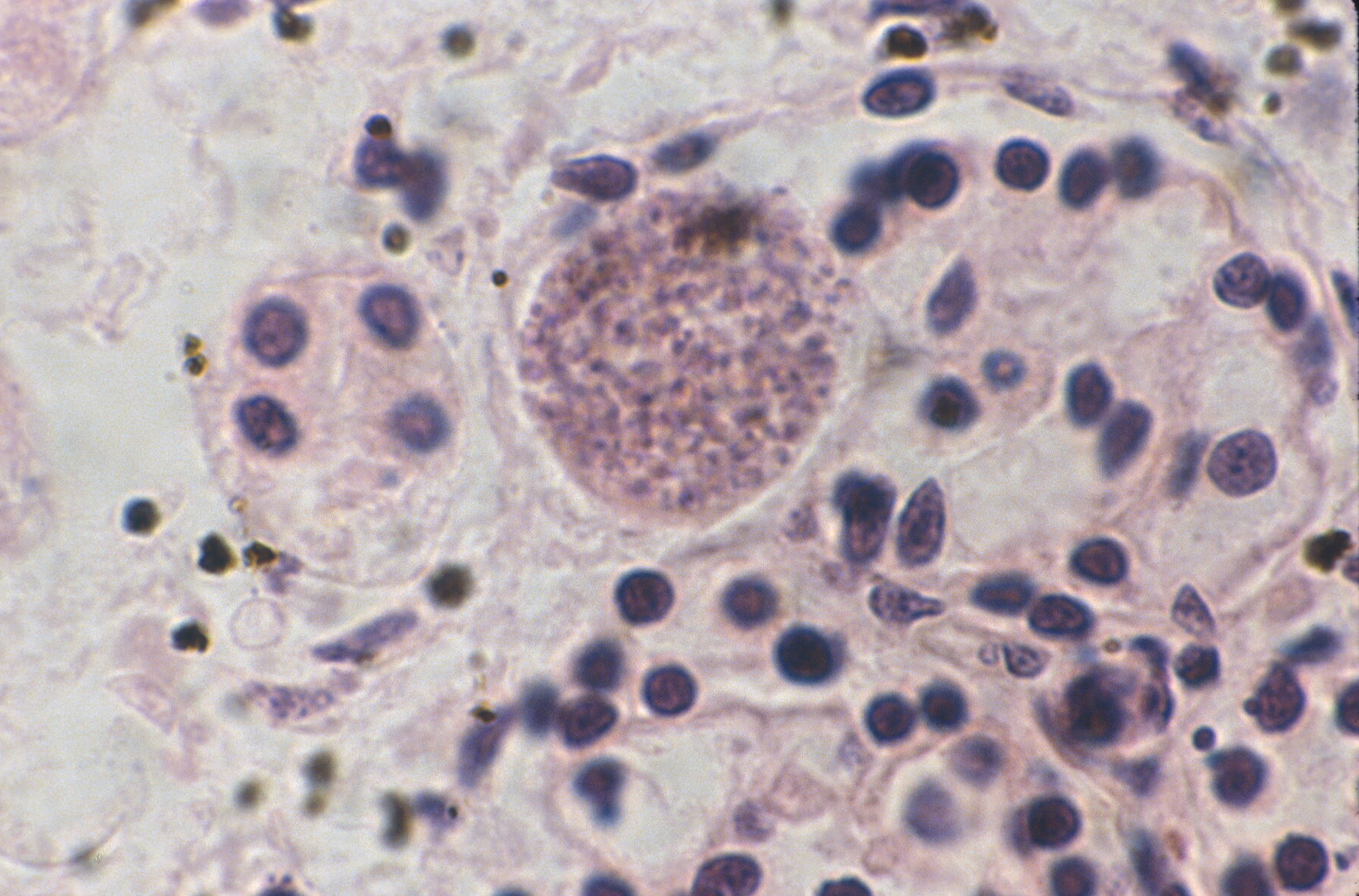
Toxoplasma cysts may be trapped in glomeruli (fig 21).
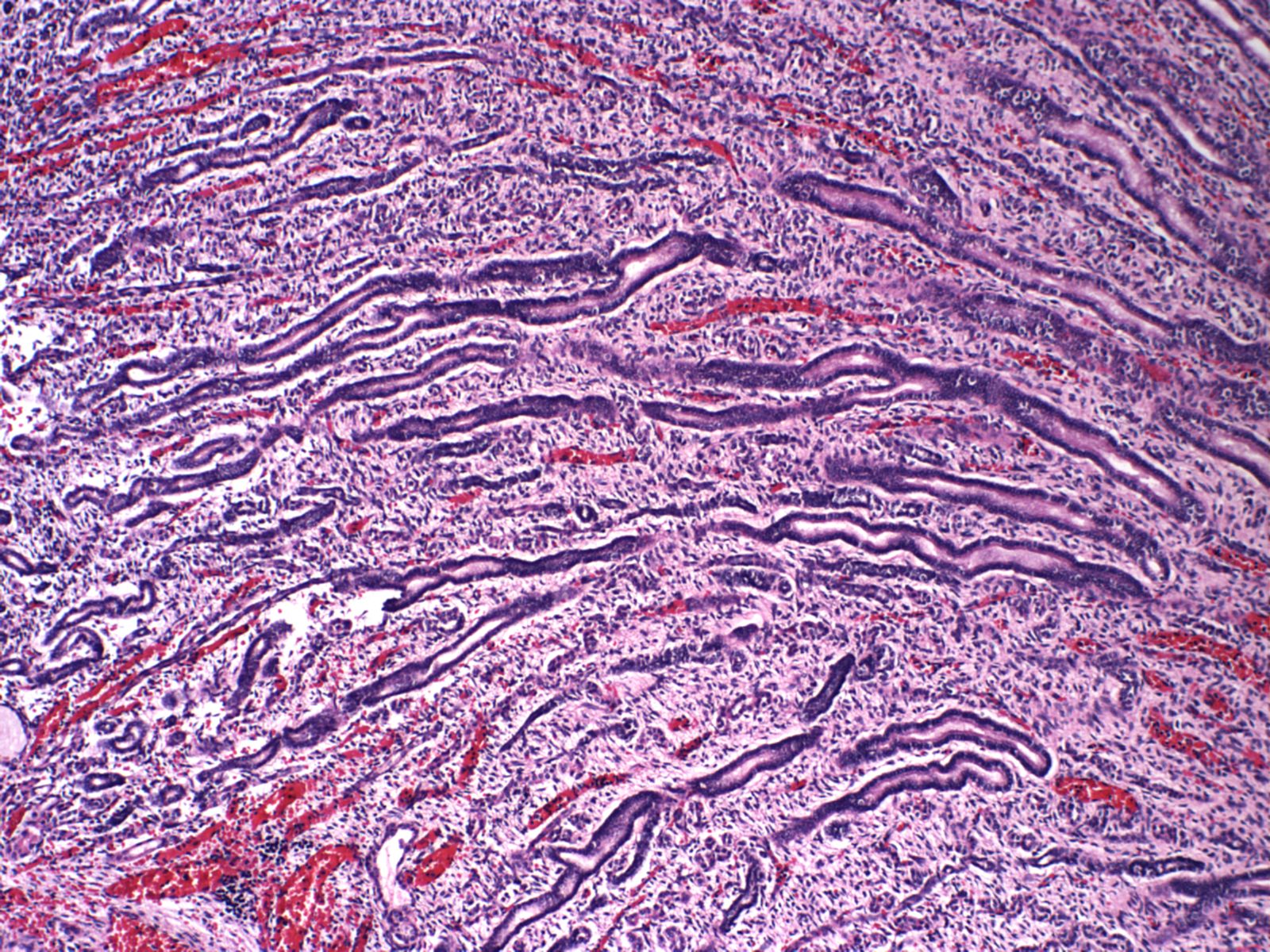
Extramedullary hematopoiesis: (Fig 22)

Supplement “A” on Renal Tubular Dysgenesis (RTD)
I have reviewed only the papers in my file, see below, but I find no attempt in the published medical literature to distinguish genetic from environmental causes of renal tubular dysgenesis on the basis of the completeness of loss of proximal tubular differentiation. There is no logical basis to do so. The lesion even in congenital cases is believed to be the result of ischemia of the glomeruli, and has been reported for example by electron microscopy to show some evidence of proximal tubular differentiation. RTD is not a primary failure of differentiation of the proximal tubules. Such a hypothetical disease would probably produce polyhydramnios. The microdissection studies in presumed inherited cases showed loss of efferent arterioles, and constriction if not separation of the neck of the connection between proximal tubules and Bowman’s space. Interestingly this is the area of the juxtoglomerular apparatus and of renin production. The ability of ACE inhibitors to cause renal ischemia may be the result not only of global ischemia but somehow, as with the skull hypoplasia, of a local effect as well. There is no a priori reason to think an inherited mechanism would produce a more complete lesion than a teratogenic mechanism.
Summary of reviewed articles
1983: Two stillborn infants (34 week, female; 36 week female) of a single non-consanguineous family with multiple anomalies, but with absent proximal tubules and all tubules “appeared abnormally developed, primitive, and reminiscent of collecting tubules”[3]. The photomicrographs resemble those of cases that will be called RTD. The kidneys were described as normal size but with loss of the cortico-medullary junction.
1985: two families of consanguineous parents. One case from one family( 32 week, male), two from the other (32 and 36 week, both male) (latter family also had female with olioghydramnios syndrome and horseshoe kidney but had proximal tubules)[4] (They called it hypernephronic nephromegaly with tubular dysgenesis).
This study microscopy: “ apparent increase in the number of glomeruli. The tubules appeared immature and hypoplastic, and proximal and distal convoluted tubules could not be differentiated. The tubules were separated by increased amounts of interstitium composed primarily of undifferentiated mesenchymal tissue.”
They also did microdissection studies that showed the tubules were often not connected to glomeruli, and glomeruli often had only an afferent, with no efferent arteriole. Papillae had decreased pores, average 2.
This study suggested X-linked or possibly autosomal recessive transmission, reasonably based on family trees of consanguinuity and multiple fetal losses.
1994: Tissue from one of the consanguineous families from 1985 was stained with lectins and compared to numerous cases of acquired renal ischemia[5]. That study concluded: “ The renal cortical tubular staining patterns observed in this study in infants with congenital renal tubular dysgenesis (RTD) or comparable tubular atrophy of early postnatal cause and in older children with end-stage renal disease with renal cortical atrophy were in general sufficiently similar to suggest that ischemia of renal tubular parenchyma is of pathogenetic importance in all these categories.” They did suggest that congenital cases would have different developmental features, but this was not referring to the tubular staining.
1986: Two siblings (36 week, male; 34 week, female) with lethal oligohydramnios sequence from non-consanguineous parents. Mother did have a cocaine overdose with one of the pregnancies[6]. The first infant had marked oligohydramnios but the second had amniotic fluid at the lower limit of normal. The kidneys were of normal weight with normal gross architecture. This report demonstrated disorganized tubules with primitive epithelium that stained for EMA (proximal tubule epithelium normally does not). There were increased glomeruli and interstitial tissue. The low power photomicrographs demonstrate a different appearance from the above cases with dilated tubules, less Bowman’s space dilatation, and fewer more mature glomeruli.
1988: Case report (32 week male) reported a case with striking feature being increased collecting ducts between glomeruli. “..only rare segments identifiable as proximal convoluted tubule.[7]” Microphotographs demonstrated yet a third slightly variant pathology.
1989: Report of 4 siblings with pathology of 2 (35 week, female; 32 week male)[8]. One had somewhat subjective malformations but other had widely patent fontanels. There was renal histology on all four which showed lack of proximal tubular differentiation and apparent crowding of glomeruli. Glomeruli occassionaly had arterioles visible on the section. The tubules were negative for EMA, Pas, and winged pea lectin which would have identifies proximal tubules although on electron microscopy one did show rudimentary brush borders. The photomicrograph shows dilated irregular branching tubules.
1990: Report (34 week, male) with severe oligohydramnios noted by 27 weeks, mother on enalapril whole pregnancy due to severe SLE related hypertension[9]. Grossly normal to mildly enlarged kidneys with histology of paucity of tubules, increased immature glomeruli. The photomicrographs look identical to those in 1983 report.
1991: A search of 500 perinatal autopsies from 1981 to 1985 (likely before much use of ACE inhibitors) for kidneys without proximal tubules[10]. Found one already published case, 4 very premature moonochorionic twin pregnancies (1 twin in each pair), and a hydropic trisomy 21 infant with hydrops and oligohydramnios. Three twins were presumed twin to twin transfusion, and the other an acardiac. The findings were interpreted as supporting the concept that the loss of proximal tubules was a consequence of ischemia.
1991: Two more cases of renal tubular dysplasia of infants of mothers on ACE inhibitors (33 week female, and 32 week male that survived renal biopsy at 1 month)[11]. These cases also had skull hypoplasia. The lethal case had bilaterial renal vein and inferior vena cava thrombosis. The photomicrograph demonstrates a typical appearance and the caption states “ “There is poor to no differentiation between proximal and distal convoluted tubules, and intertubular mesenchyme appears to be increased.” The photograph of the biopsy states only “…and poor differentiation between proximal and distal tubules.” In summary the authors state “In the ACE inhibitor-exposed cases, the renal lesion is similar to, if not identical with, that seen in both the genetic recessive form of tubular dysgenesis and ischemic renal injury.” They speculate that the hypocalvaria may be due to ischemia as membranous bone formation requires high oxygen tension, versus some receptor similarities between the action of ACE on the efferent arteriole and its action on some bone morphogenic hormone.
1992: A multiple disease comparison paper which the 1990 enalapril case with the cases above and two mothers on Aldomet, and 2 twin to twin confusions and various controls[12]. They conclude that the twins show loss of proximal tubules, like the ACE inhibitory death in the above 1991 report, and that the 1990 enalapril case and one of the Aldomet treated infants both had loss of distal tubules. They use no special stains and only publish low power photomicrographs.
1992: Report of nine further cases with very little detail beyond documentation of oligohydramnios and renal pathology[13]
1993: 3 siblings (38 week male, 20 week female, 21 week male) with the oldest gestation showing hypocalvaria, renal tubular dysgenesis and bilateral renal vein thrombosis. The two early gestation siblings were aborted because of severe oligohydramnios[14]. In the two younger gestation fetuses there were fewer proximal tubules and those present had less cytoplasm than gestation matched controls. The mother’s only medical history was a Bell’s palsy treated with prednisone in the third trimester.
1993: 3 cases of ACE inhibitor exposure[15]. Renal biopsy at 4 weeks of age demonstrated “lack of differentiation between proximal and distal tubules” and autopsy in another case states “ minimal differentiation of proximal convoluted tubules. (This case was figure 3 which was too dark for me to evaluate)
1994: A renin immunoperoxidase study of the two cases in reference 6 as well as a new case (33 week female) demonstrated increased rennin staining in intensity and distribution compared to controls[16]
2000: A study of 8 infants with renal tubular dysplasia using lysozyme staining as a marker of proximal tubular brush border[17]. The results showed that staining was absent in 4 infants and reduced in 4 others. The paper does not discuss whether the lesions could have been secondary to ACE inhibitors.
2003: A report of a case of renal tubular dysplasia in a hydropic trisomy 21 fetus that also includes a review of the literature[18]. While my review does not cover all of the cases and intentionally skipped cases related to twins or NSAID’s, the discussion begins “The histologic diagnosis of RTD depends on demonstration of marked decrease to complete absence of proximal renal tubules.”
Supplement “B” on Oligohydramnios
Oligohydramnios sequence (Note this supplement ends with its own reference list that did not mesh with Endnote from the main text or the RTD supplement)
Summary
Oligohydramnios literally means little water in the amnion. In lethal cases, there is almost no fluid during middle and late gestational development of the fetus. This has three main effects. First, the uterine wall compresses the fetus preventing movement and deforming the face and limbs. This produces a variety of limb deformities and a Potter’s facies, an appearance likened to putting a stocking cap over the face. Second, there is no longer a continuous column of fluid to transmit the pressure from the uterine wall to the bronchial tree. This pressure is necessary for lung growth, and without it pulmonary hypoplasia results. Third, there is loss of lubrication of the skin and amnion surfaces resulting in intertriginous excoriations and amnion nodosum. The extent of these effects will depend on the time of onset and the severity of the decreased fluid. In some cases, there may only be subtle deformations and little else to externally mark the oligohydramnios.
The diagnosis of oligohydramnios sequence can be made prior to the autopsy from the prenatal ultrasound, a history of no fluid with rupture of membranes and the physical features of the infant. Pulmonary hypoplasia usually results in the death of live born infants within the first 24 hours of life. Attempts to ventilate the infant often necessitate high airway pressure that causes air leaks (pneumo- thorax, mediastinum or pericardium).
Oligohydramnios is a risk factor for stillbirth in late gestation pregnancies (1) (2). There may be a disproportionate number of earlier gestation stillbirths with oligohydramnios, including those with urinary tract obstruction (3). The most plausible explanation is that the umbilical cord is more susceptible to compression or kinking if there is no surrounding fluid.
Autopsy considerations
The correlation of the degree and cause of prenatal oligohydramnios with the fetal features
The prenatal diagnosis of severe oligohydramnios often results in an elective termination of the pregnancy. There are no simple criteria for determining which second trimester features would not have survived after term delivery. The anatomic severity of the oligohydramnios sequence can be correlated with the ultrasound evaluation of the severity and duration of decreased fluid, and with the underlying diagnosis. The underlying diagnosis is important, for example absent kidneys in all but monoamnionic twins is lethal. Diseases permitting some amnion fluid accumulation or developing anhydramnios only late in gestation may not always result in neonatal death. Survival may depend on the lung volume being adequate for the gestation at delivery. For example, if anhydramnios occurred suddenly at 27 weeks gestation with cessation of lung growth, the lungs may be able to support respiration until delivery at 30 weeks, but not at 38 weeks of gestation.
Amnion nodosum may also provide a measure of the severity of oligohydramnios in older gestation infants. It is not usually a feature of oligohydramnios from prolonged rupture of membranes in which urine may be periodically wetting the amnion cavity. Late gestation onset anhydramnios appears to have a finer gross texture than earlier onset. Figure (X) demonstrates a case of very delicate amnion nodosum and moderate renal dysplasia from an infant with oligohydramnios from posterior urethral valves. He required days of high pressure ventilation but survived, although a creatinine above one predicted that he would experience later renal failure. The size and thickness of the amnion nodules contrasted markedly with the much coarser nodules found at similar gestation with absent kidneys. The inference was that there had been adequate urination until some time in the third trimester.
Accurate description and photography of the features of amnion nodosum correlated with the underlying disease and the findings of prenatal ultrasound can provide insight into the affects of different patterns of oligohydramnios. A database of such correlations will be useful in evaluating the effects of therapeutic attempts at reversing oligohydramnios (4).
The mechanism of the oligohydramnios
Fetal amnion fluid is the difference in volume between fetal urination and fetal swallowing of amnion fluid. The lung does excrete fluid, especially in the experimental situation of an externalization of tracheal secretion, but it does not appear to make a substantial contribution to normal amnion fluid volume. A two-way flow of water may also occur between the chorionic surface vasculature and the amnion cavity. Despite these potential fluid sources, starting in the second trimester, there is no amnion fluid if there is no urine. This conclusion leads to a simple branching approach to the mechanism of oligohydramnios. The first branch is between renal and non renal causes. Of the latter, the only significant mechanism is rupture of the fetal membranes and related membrane defects. The renal causes can be subdivided into decreased renal perfusion [19], intrinsic renal disease and urinary tract obstruction [19]. Prerenal oligohydramnios may be seen with severe utero-placental ischemia and with chronic twin to twin transfusion syndrome in the donor. The intrinsic renal causes are primarily ageneisis, dysplasia, cystic diseases and renal tubular dysplasia. Urinary tract obstruction is the most common mechanism of oligohydramnios sequence and can occur at different levels of the urinary tract, with the urethra being the most common. Each branch of this scheme will lead to consideration of specific etiologies.
Intrauterine growth restriction:
Growth restriction is often included as a feature of the oligohydramnios sequence. In one series of non-syndromic cases, fetal weight was below the 10th percentile in 17 of 62 cases (27%) (5). Severe uteroplacental ischemia and the donor in twin transfusion syndrome are both associated with restricted growth and oligohydramnios. In such cases, the small size is not a consequence of the oligohydramnios, but rather both are the consequence of the underlying disease. Fetal growth restriction has been observed in experimental models of oligohydramnios (6). A single research abstract reports that in nephrectomized sheep, those with continuous intra-amniotic saline infusion had normal weights, but those without fluid were growth retarded (7). Such a direct effect of fluid could be from decreased muscle mass from restricted fetal movement or from the lack of recycled nutrients and fluid from fetal swallowing. Growth restriction is at least a biologically plausible consequence of oligohydramnios.
Growth restriction is usually diagnosed post mortem if the fetus is small for gestation. Reduced mass of kidneys, lung and bladder, could make some appropriately grown infants measure small for gestation. More often megacystis, or large cystic or cystic-dysplastic kidneys would inflate the birth weight of the infant, masking growth restriction. A better post mortem measure of intrauterine growth in such cases would be femur length or mid thigh and mid calf circumferences.
Olioghydramnios as a cause of intrauterine brain injury
If the hypothesis that oligohydramnios predisposes to fetal death from umbilical cord compression is true, then is reasonable to postulate that less than lethal cord compression could cause brain lesions. An autopsy study demonstrated a significant correlation of ischemic white matter injury with oligohydramnios, but the same study also found a significant correlation with growth retardation (8). Since the two factors were not compared, a independent role for oligohydramnios could was not established.
The brain lesions described in two series of “Potter syndrome” demonstrated, without control cases, predominately minor or subjective lesions, e.g. persistent radial appearance of the cortex, under rotated dentate and cerebellar heterotopias (9) (10). One infant with dysplastic kidneys had white matter necrosis (10). Amnio-infusion in a case of renal agenesis maintained lung growth, but resulted in large cortical white matter cavities (11). The evidence is sufficient to warrant careful neuropathology in all cases of oligohydramnios.
More detail
Dr. Edith Potter in 1946 first described pulmonary hypoplasia and distorted facial features in 20 anephric infants (12), a combination that became known as Potter syndrome. Drs. Bain and Scott who reviewed 50 patients with oligohydramnios from varied lesions of the urinary tract (13) added amnion nodosum, talipes (which had been noted in Potter’s cases) and spade like hands to the inventory. Subsequent papers noted the similarity to Potter’s cases of infants with prolonged amnion fluid leakage (14). Amelioration of the features of Potter syndrome was reported in an anephric infant who was a monoamniotic twin (15). Thomas and Smith synthesized these observations into the oligohydramnios tetrad concept (“the altered facies, aberrant hand and foot positioning, late fetal growth retardation, and pulmonary hypoplasia”) (16). Dr. Potter criticized the concept that “the epicanthic fold, malformation of the tragus and antitragus, the abnormally low slanted position of the ears, the changes in subcutaneous tissue [resembling dehydration], the growth deficit, the increased frequency among boys, the absence of the uterus in girls, and the high frequency of additional malformations” could be a “direct result of oligohydramnios”. She suggested the use of the term “renal nonfunction syndrome” (17), but the Thomas and Smith concept prevailed. The oligohydramnios tetrad was replaced in Smith’s terminology with oligohydramnios sequence, i.e. a sequence because all the anomalies can be explained on the basis of a single problem in morphogenesis that leads to a cascade of subsequent defects.
The understanding of the relationship of pulmonary hypoplasia to oligohydramnios still has gaps. Multiple reports have confirmed that normal lung volume occurs in monoamnionic twins in which one twin has an anomaly that should have produced lethal oligohydramnios (18) (19). The other cases were primarily VATER association and caudal dysgenesis rather than absent kidneys. The mechanism for lung development could have been either the lack of transmitted intrauterine pressure, or a growth factor present in the urine.
An experimental study in lambs demonstrated that chronic externalized drainage of fetal pulmonary secretions results in pulmonary hypoplasia (20). The experiment was not definitive because a urine growth factor in urine would have been swallowed, but not aspirated into the lung. The same experiment demonstrated that tracheal ligation produced enlarged lungs, a finding also evident in human tracheal and laryngeal atresia (21, 22) (6). This result argued that lung growth did respond to pressure in these cases from obstructed efflux of lung secretion (23). An in-vitro study demonstrated that stretch stimulated rat lung epithelial cell proliferation. Creating drainage of the amniotic fluid into the peritoneal cavity is a common experimental model of producing oligohydramnios and pulmonary hypoplasia (24)(25). In the lamb, pressure measurements demonstrated that tracheal pressure rose above amniotic pressure, and lung fluid volumes were reduced (26) . Intra-amnion pressure measurements in humans with oligohydramnios also demonstrated decreased pressure. Direct experimental demonstration of the role of pressure was achieved by producing pulmonary hypoplasia in rabbits by cutting an aperture in the uterine wall that reduced measured amniotic pressure (27). In this model, any urine growth factors would still have been ingested. Nicolini et.al. refined the hypothesis by noting that lung fluid egress during respiration would be less than lung fluid production at normal amnion pressures, but exceed production at the amniotic pressure found with oligohydramnios (4). The complete hypothesis is that the trachea to amnion pressure gradient controls lung growth via the flow of fluid with the likely consequence that decreased lung fluid volume decreases distal airway stretch. Such a mechanism would adjust pulmonary growth to fit the pleural cavity, for example mass lesions or a constricting thorax would produce an increased intrathoracic pressure and hence increased trachea to amnion pressure gradient.
A side issue is whether a brief decrease in amnion pressure from amniocentesis can cause abnormalities of lung growth. Because of a clinical assessment of increased respiratory distress in infants who had undergone amniocentesis, a small study was done in monkeys demonstrating pulmonary hypoplasia with early gestation, but not late gestation amniocentesis (28). The decreased lung volume was not quantified in the report, and the affected monkey infants also had a lower birth weight. Another study measured crying capacity in infants with and without amniocentesis and found a decreased volume in the amniocentesis group (29). Both studies had only small numbers of subjects and most small effects, but autopsy series with relatively large numbers of cases might be able to demonstrate a difference in lung weight with amniocentesis.
Autopsy database
The documentation of the features of oligohydramnios overlaps with that of the features of the individual features causing the condition. The database for oligohydramnios will reflect some of the overlap, but the details specific to a disease will be covered thoroughly in the individual sections
Clinical history
Prenatal ultrasound:
The onset and quantification of oligohydramnios
Amnion fluid is usually measured as an index based on the amniotic fluid index, i.e. total linear measurements in four quadrants, or as a measure of the largest pocket of fluid (30). Oligohydramnios is usually defined as the largest pocket less than 1 cm or as an amniotic fluid index ≤ 5 cm or the fifth percentile for gestation. Based on amnioinfusion at term, a 4 cm index is approximately 250 ml of fluid (31).
The associated fetal anomalies
Anhyramnios can obscure the ultrasound image, but the basic information will most likely be recorded including the number of kidneys, their echogenicity (which is increased in some cystic diseases), the presence of any renal cysts, and a measure of any utero-pelvic junction dilatation. Dilated bladder or ureters should be recorded. An error that occurred in the past was to mistake the adrenals, which are flattened and displaced caudad, for the kidneys
The features of twin to twin transfusion
The documentation of twin transfusion includes evidence of an absent or thin septum, of size discordance between the twins, and of polyhydramnios in the large twin, as well as oligohydramnios of the small twin. The onset of hydrops may occur in either twin. The effects of treatment such as repetitive amnioncentesis or laser coagulation of placental vessels should be noted.
Maternal medications:
ACE inhibitors: These drugs have been associated with renal tubular dysplasia and oligohydramnios and should not be used.
Non-steroidal anti-inflammatory drugs: These drugs cause oligohydramnios and may also be a cause of renal tubular dysplasia
Steroidal drugs: Steroidal drugs, particularly birth control pills, might effect uro-genital development.
Family history:
A family history of neonatal deaths, of Potter syndrome or of non-lethal disease especially absent or abnormalities of one kidney
Maternal and Paternal renal disease: If there is a possibility of autosomal dominant renal cystic disease in the baby, the parents may even have unsuspected disease detectable by ultrasound.
Labor history
The amount of amnion fluid with rupture of membranes
Evidence of premature rupture of membranes
Placenta:
Umbilical cord length
Features of prolonged premature rupture of membranes
Amnion nodosum: the size and character of nodules
External Examination:
Potter facies:
Limb deformities:
Intertriginous excoriations:
The abdomen:
The abdomen may reveal the prune belly phenotype, or may protrude from megacystis, massive cystic kidneys or ascites.
Urogenital tract:
Lesions that suggest urinary obstruction are most important.
Other:
Special attention should be given to ear malformations, natal teeth, aplasia of the radial ray, anal atresia, encephalocele, and polydactyly. Radial aplasia may or may not include the thumb, but is usually evident in the radial deviation of the wrist.
Radiograph
A full body radiograph can detect skeletal defects associated with various causes of olioghydramnios. In cases of urinary tract obstruction, contrast injected into the bladder can better define urethral anatomy. Air leaks from distention and tearing of hypoplastic lungs can also be documented.
Internal examination
The lungs:
Combined lung weight
Lung to body weight ratio
The cut off for pulmonary hypoplasia is .015 below 30 weeks gestation and .012 at greater gestation (32). The chest radiogram may show a small, bell shaped chest, but this is often difficult to interpret.
Chest to head circumference
A small chest circumference or chest circumference to head circumference ratio, is also evidence of pulmonary hypoplasia (33), but will be affected by subcutaneous tissue, pleural effusions, and other distortions of anatomy such as the bladder elevating the diaphragms that are difficult to adjust for.
Gross description:
The lungs appear smaller than the heart in severe pulmonary hypoplasia
Cystic adenomatoid malformation has been described with oligohydramnios. In never aerated lungs, the lesion may or may not be distinctive, but often it is pale and distinctly cystic. This is not a frequent finding, but one report has suggested that it ameliorates the other oligohydramnios features through its increased secretion of lung fluid(34).
The Genito-Urinary Tract:
Bladder weight:
This is not a commonly obtained measure, but could have explanatory value. The bladder thickens with pressure load from obstruction, but dilates with residual increased volume. The bladder will hypertrophy to excrete urine until it can not achieve the pressure needed for micuration, at which point it will dilate until pressure is transmitted via the ureters to the developing renal parenchyma, leading to renal dysplasia. The ability of the bladder to compensate for obstruction may result in an inverse relationship between bladder weight and renal the generation of nephrons at which dysplasia begins.
Bladder greatest diameter and mid wall thickness
Because of the central role of the genitourinary system in oligohydramnios, each organ is described separately grossly and microscopically, including the external genitalia, perineum, urethra, prostate and vas deferens, bladder, ureters, and kidneys. The bladder is pivotal in directly further analysis of the autopsy. If the bladder is large, urethral obstruction is suspected, and sampling urine as well as post mortem cystography may be useful. If the bladder is small, intrinsic renal disease is suspected. If the bladder is normal, growth retardation suggests a prerenal cause, but a normal or large for gestation infant most likely suffers from ruptured membranes. In the thickened bladder, diverticuli and patent urachus may be present. The bladder may appear fused with other cloacal organs or the rectum and even colon may be dilated from the diversion of urine from continuity or fistula with the bladder. In some cases, uro-ascites may be present from the escape of urine from the bladder.
Typically, the urinary tract is enlarged proximal to an obstruction. Large ureters with a large bladder suggest urethral obstruction with reflux of urine which be be uni or bilateral. Sometimes only the distal ureter is dilated. A small bladder with large ureters suggests uretro-vesicular obstruction. A small bladder with markedly dilated renal pelvis indicates obstruction at the uretero-pelvic junction.
With a large bladder, the kidneys may demonstrate the distortion and gross cysts of multicystic dysplasia. With more recent urethral obstruction, the kidneys may show only much smaller cortical cysts reflecting a later onset of dysplasia. With a small bladder the kidneys may appear absent, in which case sampling the retroperitoneum microscopically may still demonstrate some dysplastic remnants. In other cases small dysplastic kidneys may be present. Large kidneys often reflect intrinsic cystic disease which may or may not be grossly apparent. Infantile polycystic renal disease and renal tubular dysplasia may have normal appearing kidneys. In cases of intrinsic renal disease without prolonged post mortem retention, some kidney may be frozen and/or fixed for electron microscopy for potential research.
Liver:
Most liver changes with renal disease are not evidence grossly except the liver may be distinctly green.
Brain
Encephaloceles can be confirmed as defects in the calvarium after the scalp is reflected during removal of the brain. Meckel Gruber syndrome may have both an occipital and a high cervical defect. The brain abnormalities in this syndrome most often affect the cerebellum.
Microscopic Examination:
Lung:
With standard sections of the lung, pulmonary hypoplasia may demonstrate smaller lobules and a sheared appearance of the parenchyma compared to similarly sectioned gestationally matched controls. Radial alveolar counts can quantify that appearance (35).
FAD
Document the features of Oligohydramnios Sequence
The final anatomic diagnosis can be “Oligohydramnios Sequence” followed by a list of the features with an indication of severity. For pulmonary hypoplasia, the lung: body weight ratio is a reliable measure of severity if body weight is not distorted by ascites, urine or hydrops. The lung weight can also be compared to the brain weight or to the normal weight for gestation. Amnion nodosum can be graded by the approximate size of the nodules.
In cases of therapeutic abortion, documentation of lung hypoplasia provides probable evidence that the infant would not have survived a later gestation delivery. In neonatal deaths, the lung hypoplasia can be correlated with complications of ventilation. The underlying mechanism of the sequence is a second major diagnosis. Th discussion may included the expected outcome and therapy for the mechanism such as urethral atresia. Further diagnoses directly related to oligohydramnios that may be listed and are any evidence of acute asphyxia in the stillborn infant and evidence of hypoxic/ischemic brain lesions.
The etiology of the oligohydramios and recurrence risk
Each mechanism of oligohydramnios has characteristic etiologies. In large series of oligohydramnios cases, there is always an array of chromosome abnormalities (5) (36). The relationship may be coincidental or perhaps due to an overall increased risk of malformation, for example trisomy 21 and obstructive uropathy. In translocations, a specific gene alteration may be responsible, but there are too few cases to analyze. Another group of cases will have patterns of extra-renal anomalies that strongly suggest a genetic or teratologic event. If the same pattern is found in multiple cases, an inherited syndrome can be suspected, although an association of defects such as the VATER or caudal dysplasia may share non-renal malformations because of common disruptions of early development. In most cases the gene involved in oligohydramnios is unknown with exceptions such as autosomal dominant polycystic disease. In many cases, the diagnosis of genetic disease is established by family history or consanguinity. Some genetic disease such as familial renal a/dysplasia may cause either absence or dysplasia of one or both kidneys (37). In an individual infant, for example without kidneys, familial renal a/dysplasia can not be excluded morphologically. Gene knockout models in mice have on the other hand identified a large number of genes that if non-functional prevent renal development. The difficulty of inferring etiology is evident in the non-discordance of monozygotic twins for oligohydramnios, which in some cases appears due to variable penetrance of a/dysplasia (38) (15) and in others due to caudal dysgenesis presumably from the complications of splitting the zygote (19) [McNamara, 1995 #4021]. Only further research will establish which genes are defective in human disease.
Teratologic causes of oligohydramnios include medical drugs such as ACE inhibitors that cause tubular dysgenesis (39). The dose and timing of a medication, as well as potential modes of action, are often more easily established than with other environmental agents. Epidemiologic evidence can show an increased risk of urinary tract malformations with oral contraceptives used after conception, but the array of anomalies was very variable [personal communication] (40). The intrinsic complexity of developmental processes may stochastically result in a small number of urinary malformations without any pathologic process. Environmental agents may increase the chance of such a developmental perturbation rather than produce a specific maldevelopment. There is currently little direct evidence for environmental agents in the causes of oligohydramnios.
Research
1. The comparison of the features of oligohydramnios with different disease may provide some insight into the critical mechanisms in pulmonary hypoplasia and renal dysplasia. Correlating the onset and severity of oligohydramnios with the pulmonary size and morphology, particularly the effect on branching versus acinar development. Similarly the comparison of the timing of urinary tract obstructive disease, as well as apparent non-obstructive causes of renal dysplasia, can provide insights into mechanism. The comparison of drug induced tubular dysgenesis with cases of prerenal oligohydramnios and even of ischemic injury may provide insight into tubular development. Studies have been done using immunoperoxidase to look at renin producing cells in twin to twin transfusion (41), and using molecular techniques to identify apoptotic cells in renal dysplasia (42). Better understanding of the pathologic processes may allow pharmacologic intervention to preserve the lung or kidney, as well as permit more critical assessment of when to deliver an infant with oligohydramnios and potential survival.
2. With parental permission, the pooling of autopsy reports and of DNA, and possibly renal m-RNA and protein, will allow the comparison of human disease with murine models of known genetic abnormality. The best way to pool the information is a computer database with essential information that would be available to researchers. The tissue could be kept at each institution, or centrally. The researchers would not have access to the patient identification. Parents could decide if they wished to be informed of any change in diagnosis or further molecular characterization of their child’s disease based on research done subsequent to the autopsy.
In cases of human disease with chromosome translocation, further family studies to identify suspect genes may be possible. Parents could be asked for permission to make their name available to qualified researchers.
References to the oligohydramnions supplement:
1. Casey BM, McIntire DD, Bloom SL, Lucas MJ, Santos R, Twickler DM, et al. Pregnancy outcomes after antepartum diagnosis of oligohydramnios at or beyond 34 weeks’ gestation [In Process Citation]. Am J Obstet Gynecol 2000;182(4):909-12.
2. Hsieh TT, Hung TH, Chen KC, Hsieh CC, Lo LM, Chiu TH. Perinatal outcome of oligohydramnios without associated premature rupture of membranes and fetal anomalies. Gynecol Obstet Invest 1998;45(4):232-6.
3. Reuss A, Wladimiroff J, Scholtmeijer R, Stewart P, Sauer P, Niermeijers M. Prenatal evaluation and outcome of fetal obstructive uropathies. Prenatal Diagnosis 1988;8:93-102.
4. Nicolini U, Fisk NM, Rodeck CH, Talbert DG, Wigglesworth JS. Low amniotic pressure in oligohydramnios–is this the cause of pulmonary hypoplasia? Am J Obstet Gynecol 1989;161(5):1098-101.
5. Curry C, Jensen K, Holland J, Miller L, Hall B. The Potter sequence: a clinical analysis of 80 cases. Am J Med Genet 1984;19:679-702.
6. King JC, Mitzner W, Butterfield AB, Queenan JT. Effect of induced oligohydramnios on fetal lung development. Am J Obstet Gynecol 1986;154(4):823-30.
7. Lord L, McConnell C, Iwamoto H. Effects of nephrectomy (Nx) and oligohydramnios (O) in intrauterine growth and the insulin-like growth factor (IGF) system in fetal sheep. Pediatr Res 1994;35:368A.
8. Gaffney G, Squier MV, Johnson A, Flavell V, Sellers S. Clinical associations of prenatal ischaemic white matter injury. Arch Dis Child Fetal Neonatal Ed 1994;70(2):F101-6.
9. Grunnet ML, Bale JF. Brain abnormalities in infants with Potter sydrome (oligohydramnios tetrad). Neurol (Ny) 1981;31:1571-4.
10. Kadhim HJ, Lammens M, Gosseye S, Gadisseux J-F, Evrad P. Brain defects in infants with Potter syndrome (oligohydramnios sequence). Pediatr Pathol 1993;13:519-36.
11. Cameron D, Lupton BA, Farquharson D, Hiruki T. Amnioinfusions in renal agenesis. Obstet Gynecol 1994;83:872-6.
12. Potter EL. Bilateral Renal Agenesis. J Pediatr 1946;29:68-76.
13. Bain A, Scott J. Renal agenesis and severe urinary tract dysplasia A review of 50 cases; with particular reference to the associated anomalies. Br Med J 1960;1:841-6.
14. Bain AD, Smith II, Gauld JK. Newborn after prolonged leakage of liquor amnii. Br Med J 1964;2:598-9.
15. Mauer SM, Dobrin RS, LVernier R. Unilateral and bilateral renal agenesis in monoamniotic twins. J Pediatr 1974;84:236-8.
16. Thomas T, Smith DW. Oligohydramnios; cause of the nonrenal features of Potter’s syndrome; including pulmonary hypoplasia. J Pediatr 1974;84:811-4.
17. Potter EL. Oligohydramnios: Further comment. J Pediatr 1974;84:931-2.
18. McNamara MF, McCurdy CM, Reed KL, Philipps AF, Seeds JW. The relation between pulmonary hypoplasia and amniotic fluid volume: lessons learned from discordant urinary tract anomalies in monoamniotic twins. Obstet Gynecol 1995;85(5 Pt 2):867-9.
19. Sherer DM, Abramowicz JS, Smith SA, Cusson CL, Metlay LA, Woods JR. Normal lung structure and pulmonary function in a twin with agenesis of the cloacal membrane and persistent severe oligohydramnios. J Mat-Fet Med 1992;1:24-8.
20. Alcorn D, Adamson TM, Lambert TF, Maloney JE, Ritchie BB, Robinson PM. Morphological effects of chronic tracheal ligation and drainage in the fetal lamb lung. J Anat 1977;123:649-60.
21. Wigglesworth JS, Desai R, Hislop AA. Fetal lung growth in congenital laryngeal atresia. Pediatr Pathol 1987;7(5-6):515-25.
22. Wright JR, Jr., Thompson DL, McBride JA, Liston RM. Asynchronous pulmonary hyperplasia associated with tracheal atresia: pathologic and prenatal sonographic findings. Pediatr Pathol Lab Med 1995;15(1):81-97.
23. Liu M, Xu J, Tanswell AK, Post M. Stretch-induced growth-promoting activities stimulate fetal rat lung epithelial cell proliferation. Exp Lung Res 1993;19(4):505-17.
24. Symchych PS, Winchester P. Potter’s Syndrome. Am J Pathol 1978;90:779-82.
25. Nakayama DK, Glick PL, Harrison MR, Villa RL, Noall R. Experimental pulmonary hypoplasia due to oligohydramnios and its reversal by relieving thoracic compression. J Pediatr Surg 1983;18(4):347-53.
26. Harding R, Hooper SB, Dickson KA. A mechanism leading to reduced lung expansion and lung hypoplasia in fetal sheep during oligohydramnios. Am J Obstet Gynecol 1990;163(6 Pt 1):1904-13.
27. Kizilcan F, Tanyel C, Çakar N, Büyükpamukçu N, A H. The effect of low amniotic pressure without oligohydramnios on fetal lung development in a rabbit model. Am J Obstet Gynecol 1995;173:36-41.
28. Hislop A, Fairweather DV. Amniocentesis and lung growth: an animal experiment with clinical implications. Lancet 1982;2(8310):1271-2.
29. Vyas H, Milner A, Hopkin I. Amniocentesis and fetal lung development. 1982;627-8.
30. Moore TR. Superiority of the four-quadrant sum over the single-deepest-pocket technique in ultrasonographic identification of abnormal amniotic fluid volumes. Am J Obstet Gynecol 1990;163(3):762-7.
31. Strong TH, Jr., Hetzler G, Paul RH. Amniotic fluid volume increase after amnioinfusion of a fixed volume. Am J Obstet Gynecol 1990;162(3):746-8.
32. Wiggleswoth JS, Desai R. Use of DNA estimation for growth assessment in normal and hypoplastic fetal lungs. Arch Dis Child 1981;56:601-5.
33. Nimrod C, Davies D, Iwanicki S, Harder J, Persaud D, Nicholson S. Ultrasound prediction of pulmonary hypoplasia. Obstet Gynecol 1986;68:495-8.
34. Krous H, Harper P, Perlman M. Congenital Cystic Adenomatoid malformation in bilateral renal agensis. Arch Pathol Lab Med 1980;104:368-70.
35. Cooney T, Thurlbeck W. The radial alveolar count method of Emery and Mithral: a reappraisal 2- Intrauterine and early postnatal growth. Thorax 1982;37:580-3.
36. Scott R, Goodburn S. Potter’s syndrome in the second trimester-prenatal screening and pathological findings in 60 cases of oligohydramnios. Prenatal Diagnosis 1995;15:519-25.
37. Moerman P, Fryns JP, Sastrowijoto SH, Vandenberghe K, Lauweryns JM. Hereditary renal adysplasia: new observations and hypotheses. Pediatr Pathol 1994;14(3):405-10.
38. Marras A, Mereu G, Dessi C, Macciotta A. Oligohydramnios and extrarenal abnormalities in Potter syndrome. J Pediatr 1983;102:597-8.
39. Martin R, Jones K, Mendoza A, Barr M, Benirschke K. Effect of ACE inhibition on the fetal kidney: decreased renal blood flow. Teratol 1992;46:317-21.
40. Li D-K, Darling J, Mueller B, Kickok D, Fantel A, Weiss N. Oral contraceptive use after conception in relation to the risk of congenital urinary tract anomalies. Teratol 1995;51:30-6.
41. Kilby MD, Platt C, Whittle MJ, Oxley J, Lindop GB. Renin gene expression in fetal kidneys of pregnancies complicated by twin-twin transfusion syndrome. Pediatr Dev Pathol2001;4(2):175-9.
42. Poucell-Hatton S, Huang M, Bannykh S, Benirschke K, Masliah E. Fetal obstructive uropathy: patterns of renal pathology. Pediatr Dev Pathol 2000;3(3):223-31.
References:
1. Potter, E. and T. ST, Glomerular development in the kidney as an index of fetal maturity. J Pediatr, 1943. 22: p. 695-706.
2. Fujikura, T. and L. Froehlich, Birthweight, gestational age, and renal glomerular development as indices of fetal maturity. Am J Obstet Gynecol, 1972. 113: p. 627-631.
3. Allanson, J.E., J.T. Pantzar, and P.M. MacLeod, Possible new autosomal recessive syndrome with unusual renal histopathological changes. Am J Med Genet, 1983. 16(1): p. 57-60.
4. Voland, J.R., et al., Congenital hypernephronic nephromegaly with tubular dysgenesis: a distinctive inherited renal anomaly. Pediatr Pathol, 1985. 4(3-4): p. 231-45.
5. Landing, B.H., et al., Labeled lectin studies of renal tubular dysgenesis and renal tubular atrophy of postnatal renal ischemia and end-stage kidney disease. Pediatr Pathol, 1994. 14(1): p. 87-99.
6. Schwartz, B.R., et al., Isolated congenital renal tubular immaturity in siblings. Hum Pathol, 1986. 17(12): p. 1259-63.
7. Bernstein, J., Renal tubular dysgenesis. Pediatr Pathol, 1988. 8(4): p. 453-6.
8. Swinford, A.E., et al., Renal tubular dysgenesis: delayed onset of oligohydramnios. Am J Med Genet, 1989. 32(1): p. 127-32.
9. Cunniff, C., et al., Oligohydramnios sequence and renal tubular malformation associated with maternal enalapril use. Am J Obstet Gynecol, 1990. 162(1): p. 187-9.
10. Genest, D. and J. Lage, Absence of normal-appearing proximal tubules in the fetal and neonatal kidney: prevalence and significance. Hum Pathol, 1991. 22: p. 147-153.
11. Barr, M.J. and M.M.J. Cohen, ACE inhibitor fetopathy and hypocalvaria: the kidney-skull connection. Teratol, 1991. 44: p. 485-495.
12. Martin, R., et al., Effect of ACE inhibition on the fetal kidney: decreased renal blood flow. Teratol, 1992. 46: p. 317-321.
13. Allanson, J.E., et al., Renal tubular dysgenesis: a not uncommon autosomal recessive syndrome: a review [see comments]. Am J Med Genet, 1992. 43(5): p. 811-4.
14. Metzman, R.A., M.A. Husson, and E.A. Dellers, Renal tubular dysgenesis: a description of early renal maldevelopment in siblings. Pediatr Pathol, 1993. 13(2): p. 239-48.
15. Pryde, P.G., et al., Angiotensin-converting enzyme inhibitor fetopathy. J Am Soc Nephrol, 1993. 3(9): p. 1575-82.
16. Bernstein, J. and L. Barajas, Renal tubular dysgenesis: evidence of abnormality in the renin- angiotensin system.J Am Soc Nephrol, 1994. 5(2): p. 224-7.
17. Moldavsky, M., A. Shahin, and H. Turani, Renal tubular dysgenesis present in a newborn with meconium ileus.Pediatr Pathol, 1994. 14(2): p. 245-51.
18. Jain, V. and D. Beneck, Renal tubular dysgenesis in an hydropic fetus with trisomy 21: a case report with literature review. Pediatr Dev Pathol, 2003. 6(6): p. 568-72.
19. postrenal., S.n.t.t.p.r.
