Pancreas
Gross Findings
Abnormalities of shape occur with poly/asplenia (heterotaxy) syndrome and with trisomy 13. An unusual anomaly, also associated with trisomy 13, is fusion of the spleen to the pancreas. (Fig 1)
Histologic sampling
If the pancreas in small, the whole organ can be submitted or if larger, cross sections of the tail, mid-portion, and head may be submitted.
Microscopic Findings
Exocrine:
Cysts, dilated and plugged ducts:
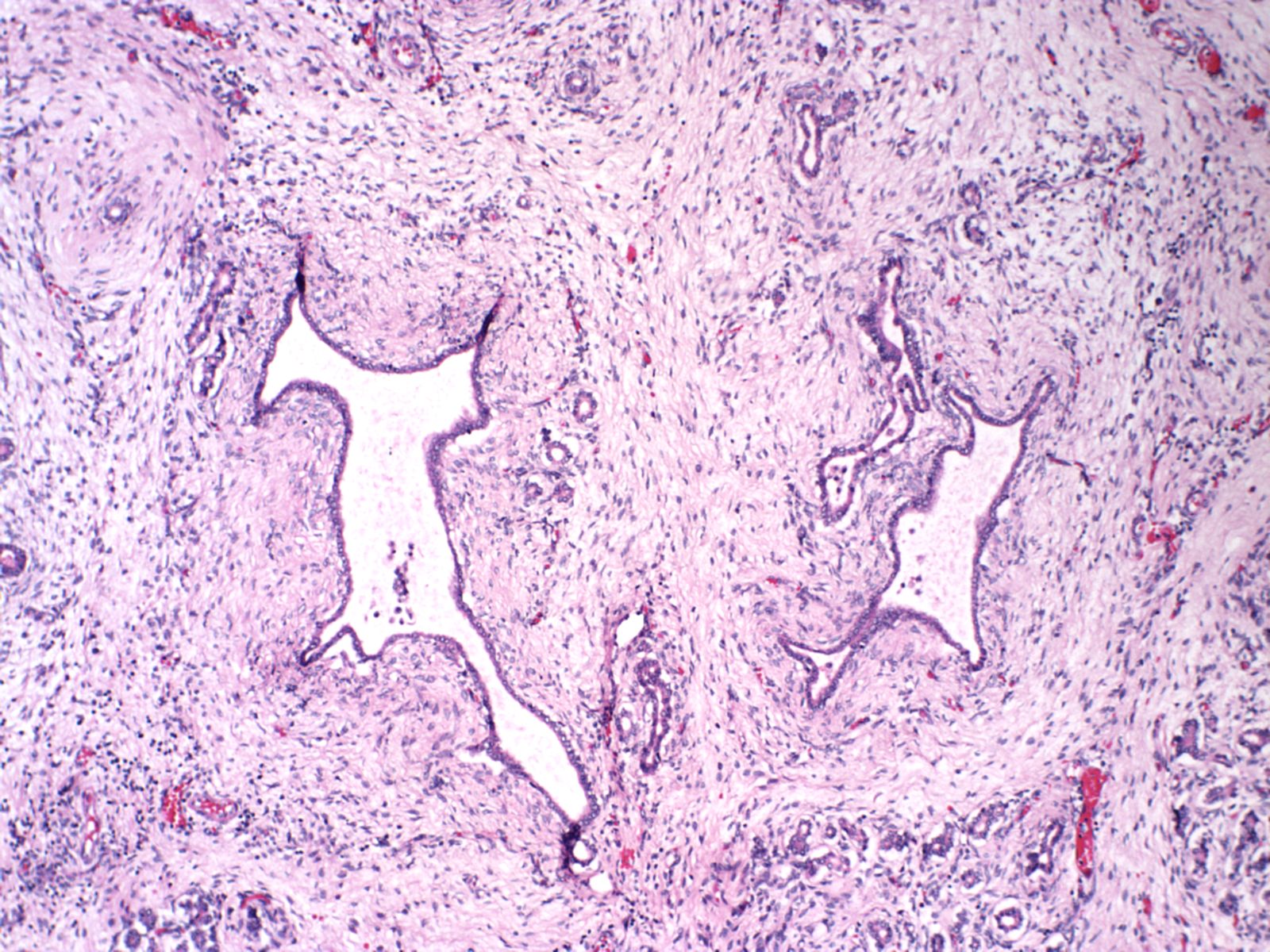
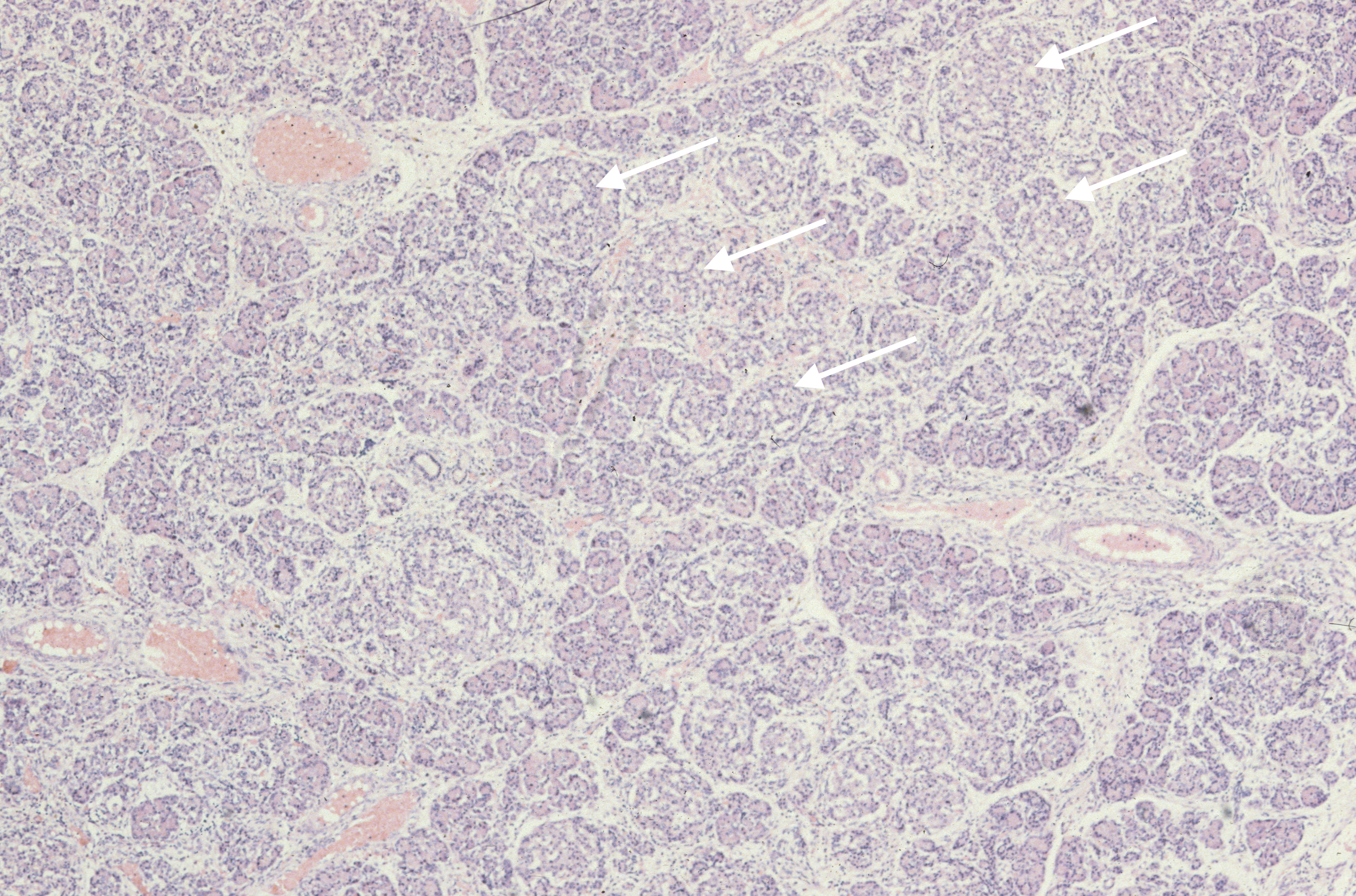
The exocrine pancreas may demonstrate malformations of the duct system such as in Ivemark syndrome of renal-hepatic-pancreatic dysplasia, Meckel Gruber syndrome, and various osteochondrodysplasias[1-3]. (Fig 2, 3) Cystic fibrosis can cause meconium ileus in utero, and may show ductal plugging in the pancreas[4]. In neonates with prolonged cachexia, often seen before total parental nutrition became available, the pancreatic ductules would be dilated with sparse secretion in the lumen.


Other:
Megakaryocytosis in trisomy 21, are associated with a similar infiltration of the liver[5, 6]. Activation of digestive enzymes and pancreatitis is rare but has been reported with congenital midgut volvulus[7]. The pancreas is often involved in more systemic processes such as increased erythropoiesis or accumulation of iron in cases of liver damage or other cause of decreased transferrin.
Endocrine:
Islet Hypertrophy, hyperplasia
Insulin
The most frequent association of islet hyperplasia and cell hypertrophy is with hyperglycemia from maternal glucose intolerance[8]. The Pedersen hypothesis states that the increased transfer of maternal substrate (glucose and amino acids) to the fetus increases fetal insulin production and results in the phenotype of the infant of a diabetic mother (IDM). The hyperplasia of pancreatic islets is a reflection of increased insulin production. The IDM phenotype includes increased fat deposition, particularly notable for fat cheeks, and cardiomegaly.
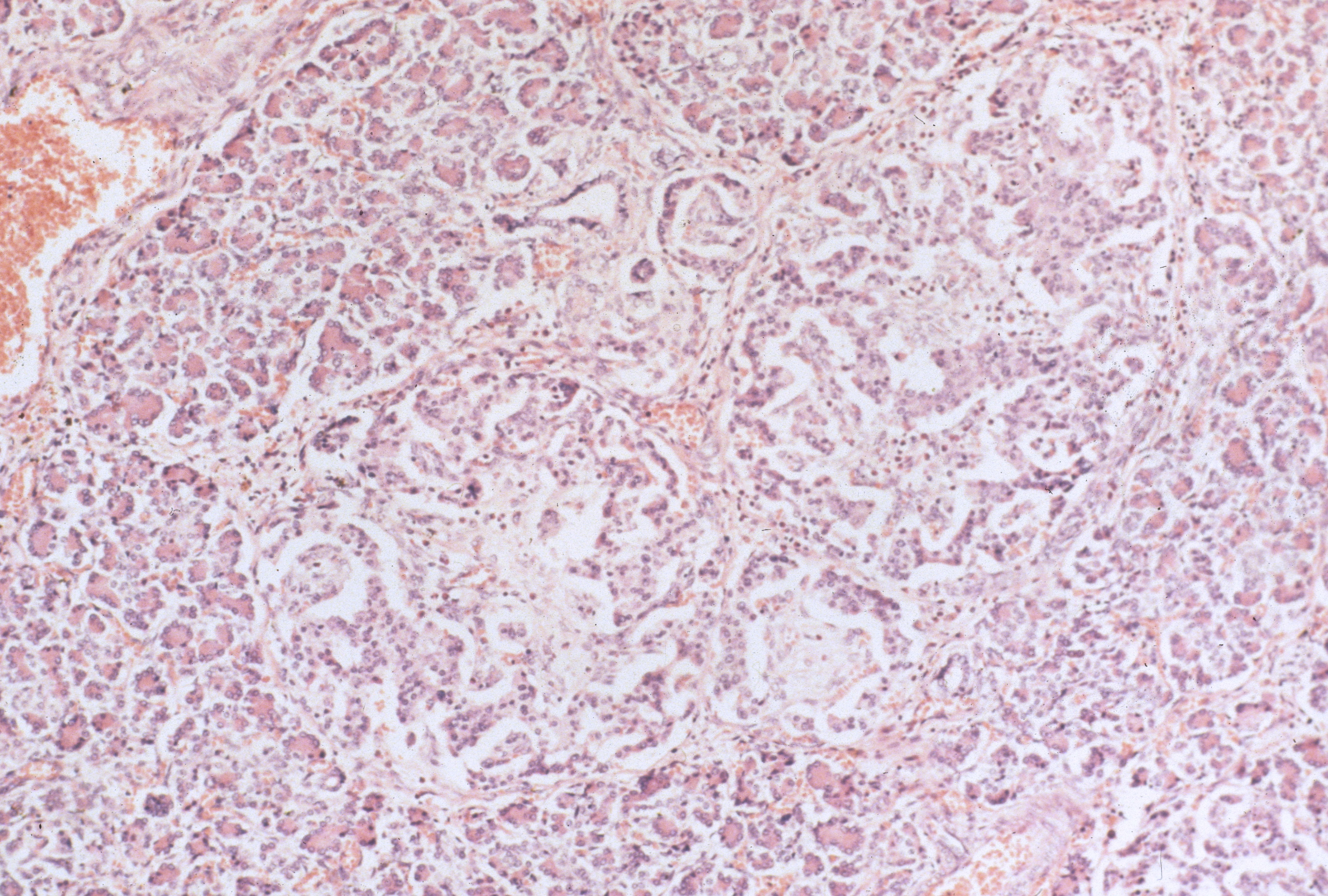
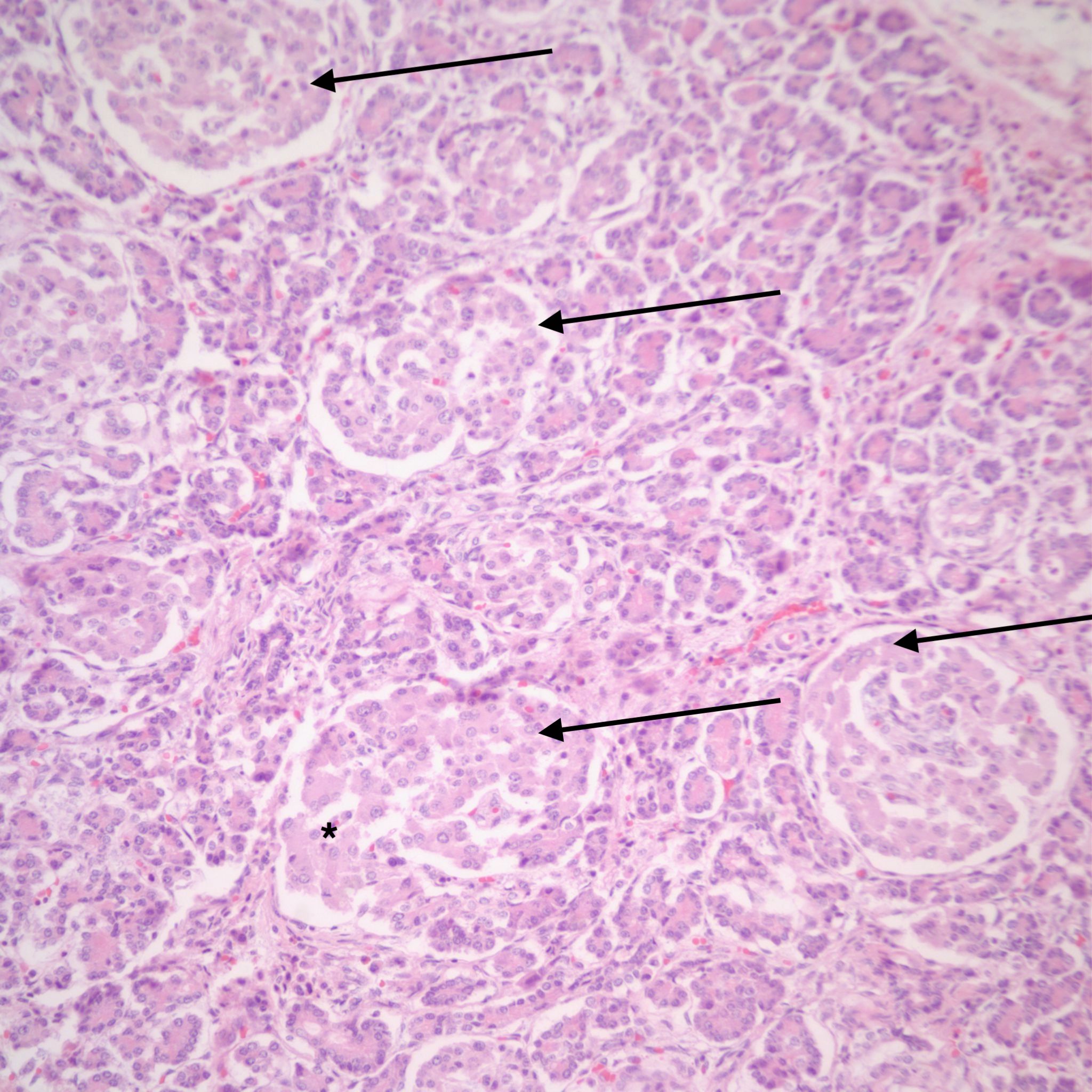
In infants of diabetic mothers, the pancreas typically demonstrates large (greater than 250 µ), plentiful islets (>7% of the pancreatic area) (Fig 5).

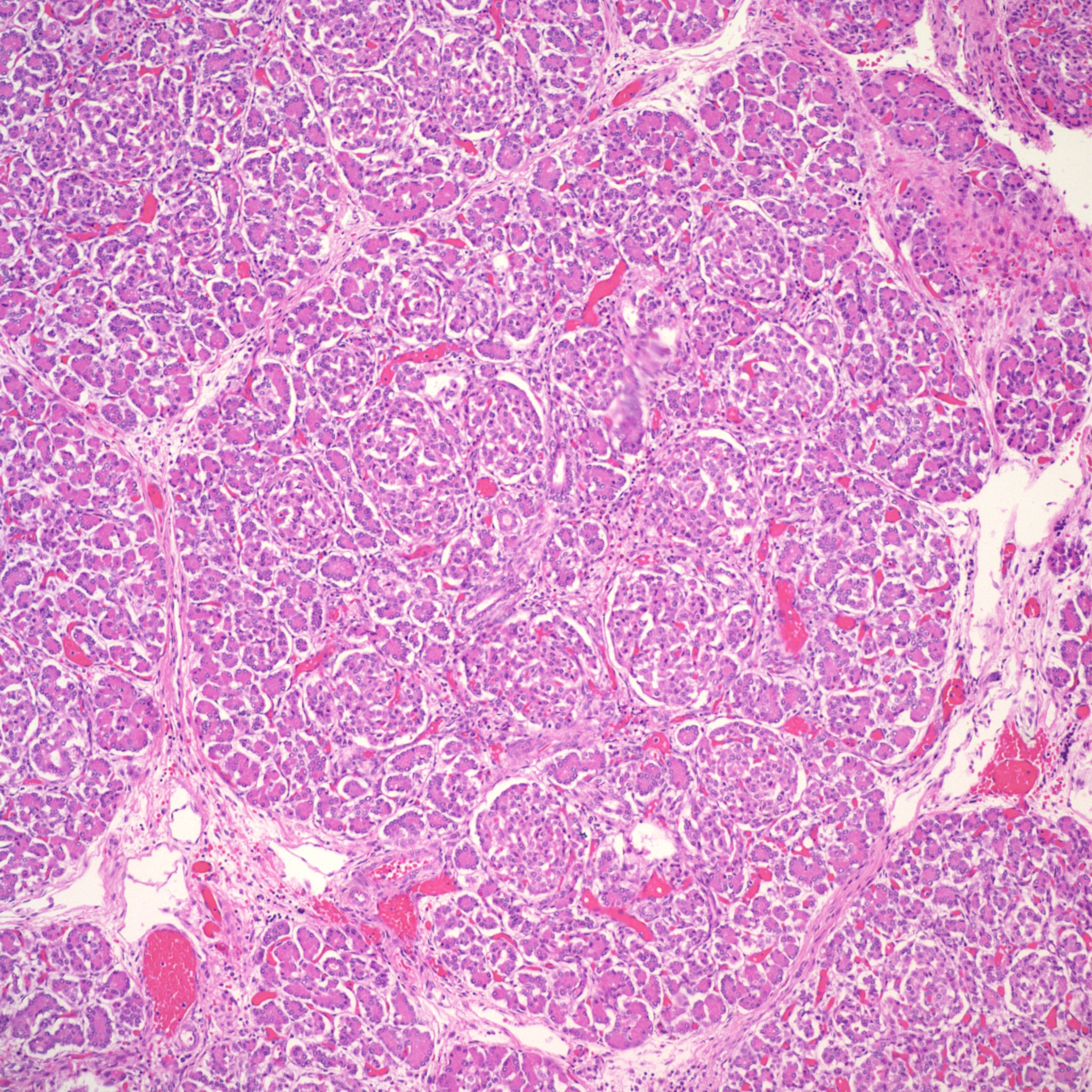
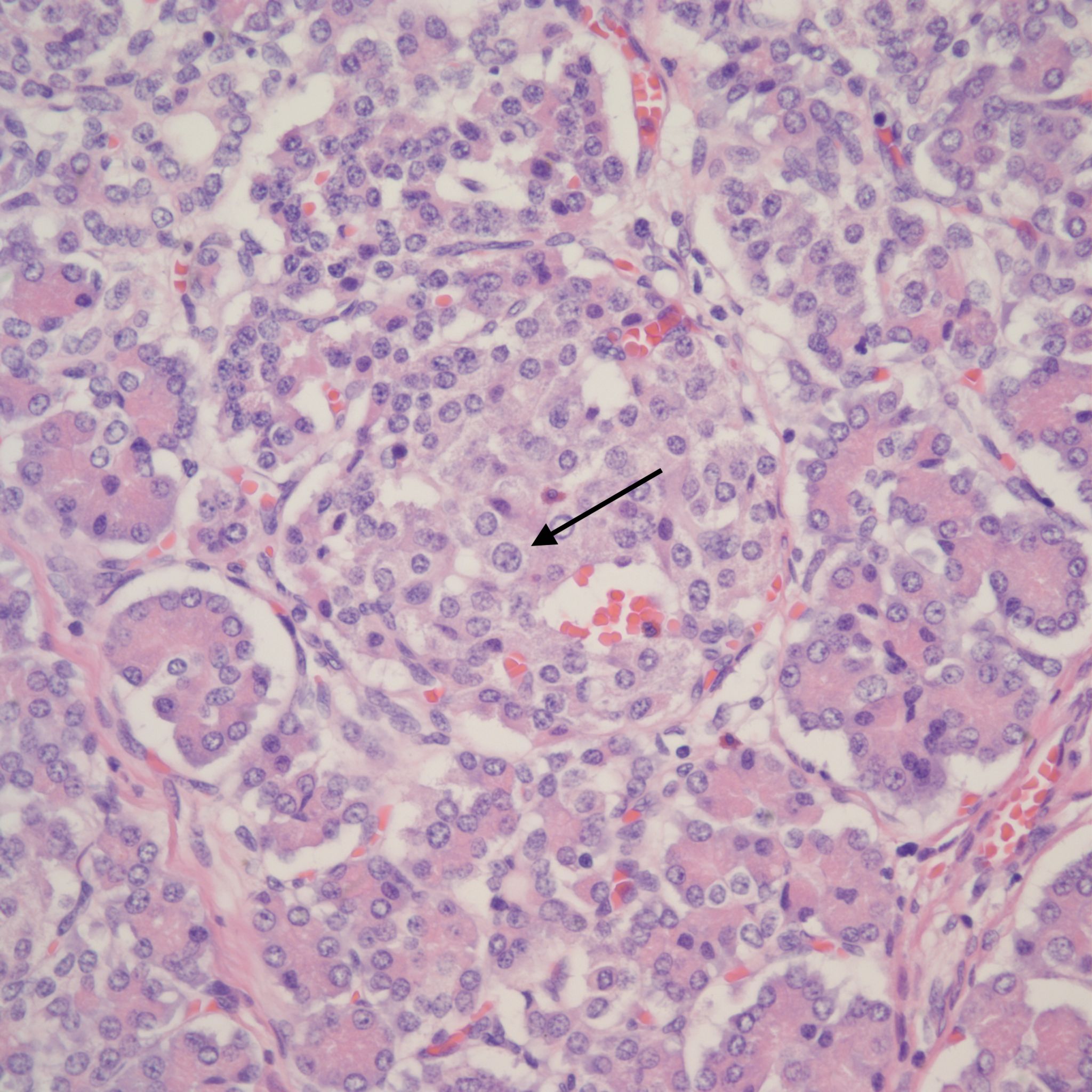
However, islet cell type and distribution in the pancreas is not homogenous throughout the organ and also changes with gestation. An increase in islet area may be difficult to determine without quantitative technique and comparison with matched controls for gestation and location within the pancreas. Beta cells that secrete insulin and amylin, normally make up greater than a majority of cells in an islet. Therefore, the percentage of beta cells in an islet is seldom helpful determining an overall hyperplasia of insulin secreting cells. A more reliable feature to identify evidence of increased islet response is cell hypertrophy. These hypertrophic cells are polyploid with clearly enlarged nuclei compared to surrounding cells (Fig 6).

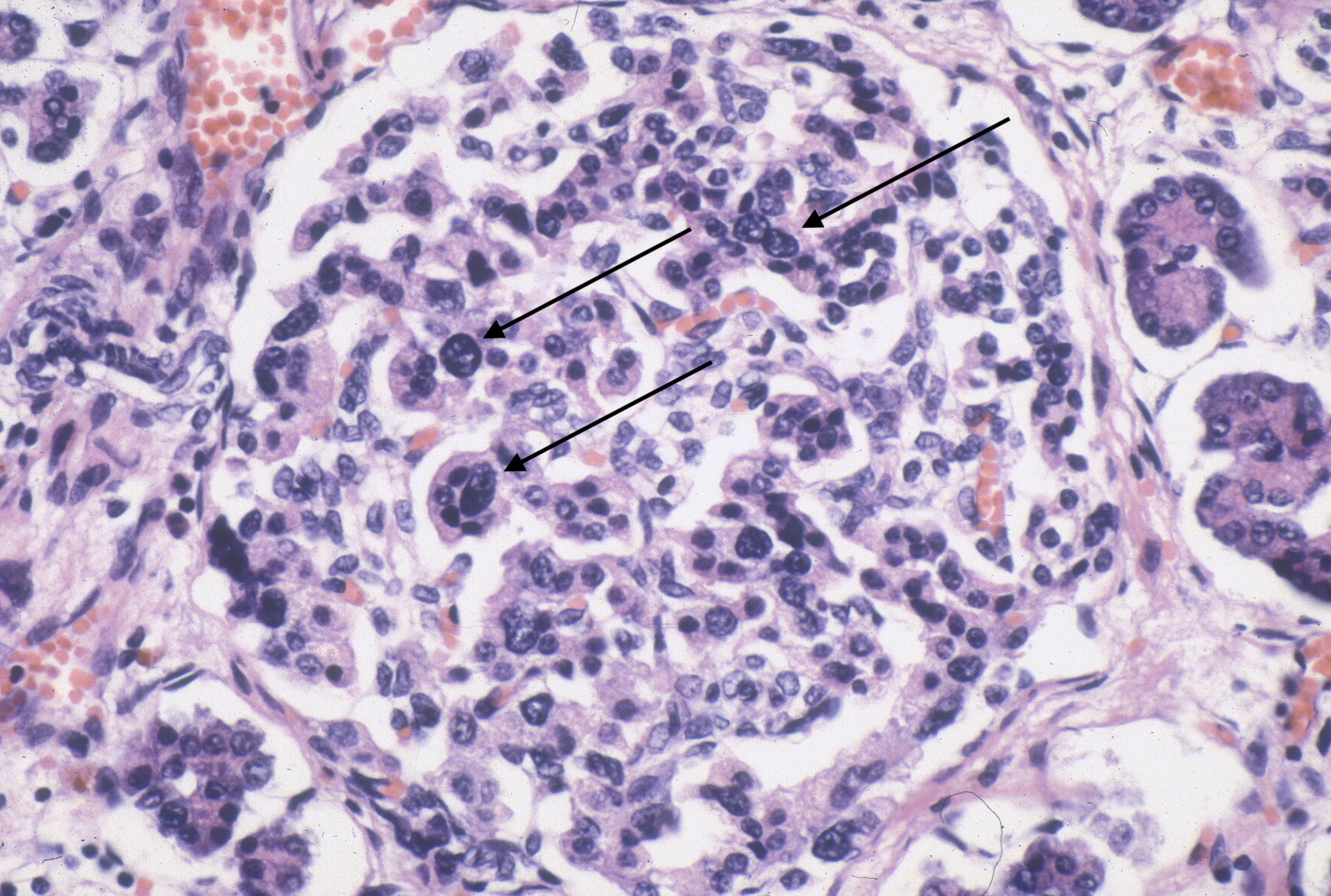
The cytomegalic cells can be identified even with marked autolysis of the pancreas (fig 7).

The hypothesis is that both hyperplasia and polyploid cell hypertrophy are adaptations to increase insulin secretion. An anti-insulin immuno-stain can confirm the identity of cytomegalic cells as insulin staining and help to visually demonstrate the size and extent of islets in general.
Van Assche has asserted that islet hypertrophy does not occur without intact hypothalamic pituitary axis[9]. This relationship does seem violated with holo-anencephaly showing the features of an infant of diabetic mother (Fig 8,9).

Macrosomia itself appears correlated with islet hyperplasia, especially in Beckwith-Wiedemann syndrome. One study looked at beta cell hyperplasia in non-diabetic, macrosomic infants[10]. Two of their ten cases demonstrated possible islet cell hyperplasia, based on larger clusters of beta cells, rather than the total area of beta cells. The criteria for the study were that the mothers be non-obese, have normal glucose screening tests, and no medications that might cause hyperglycemia. The study did not include Hemoglobin A1C levels that might have identified subtle maternal hyperglycemia.
Even without macrosomia, islet hyperplasia can be encountered in multiple clinical causes of perinatal death. Some examples are osteogenesis imperfecta, type II which is perinatally lethal (Fig 10), probable Antley-Bixler syndrome (Figs 11-14), Noonan syndrome with polyvalvular dysplasia of the heart[11] (Figs 15-17), liver disease including Zellweger syndrome, and in congenital CMV infection (Figs 18-19). On interesting case with islet cell hyperplasia had a severe caudal regression syndrome, but without maternal diabetes (Figs 20-21). Maternal hyperglycemia is associated with caudal regression malformations.
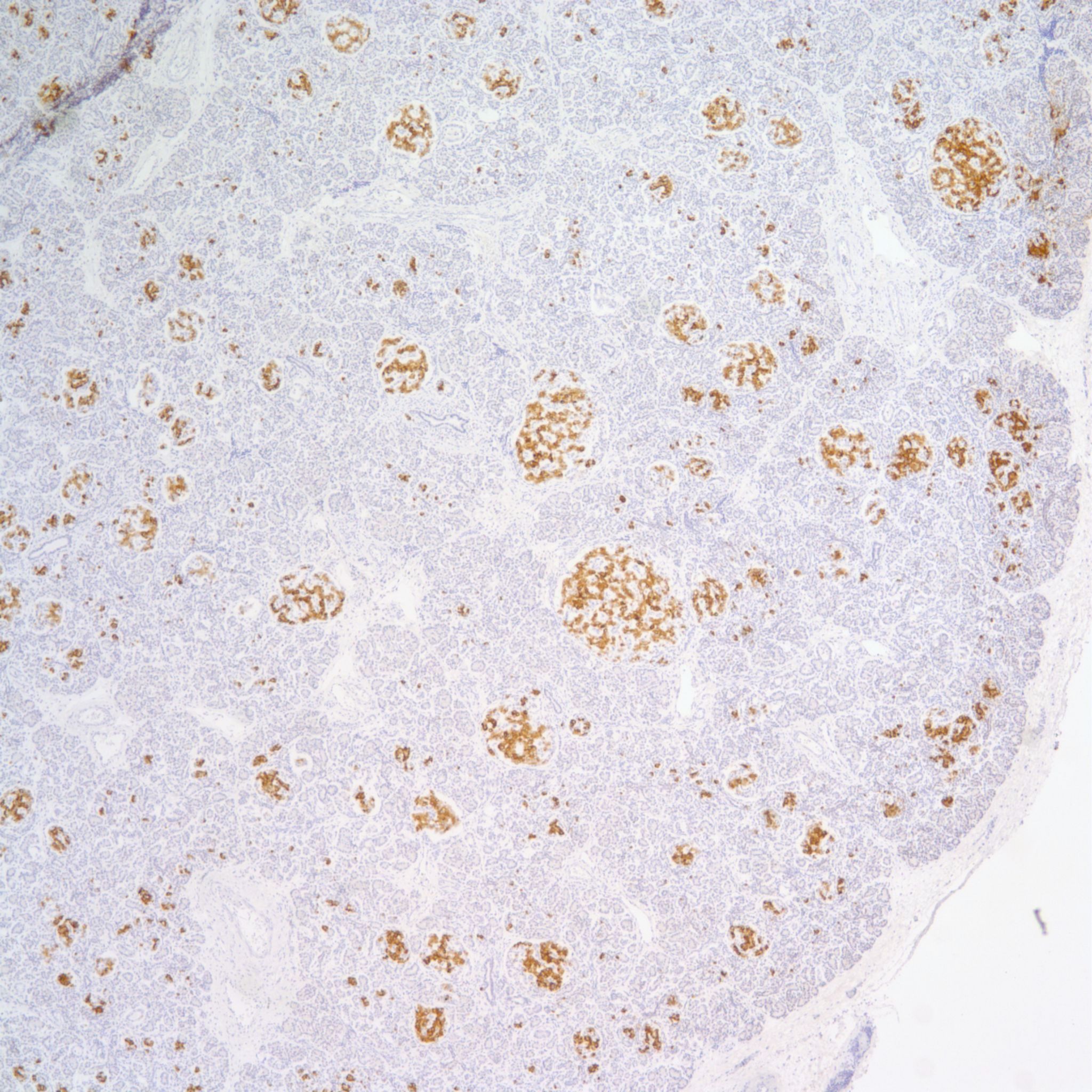



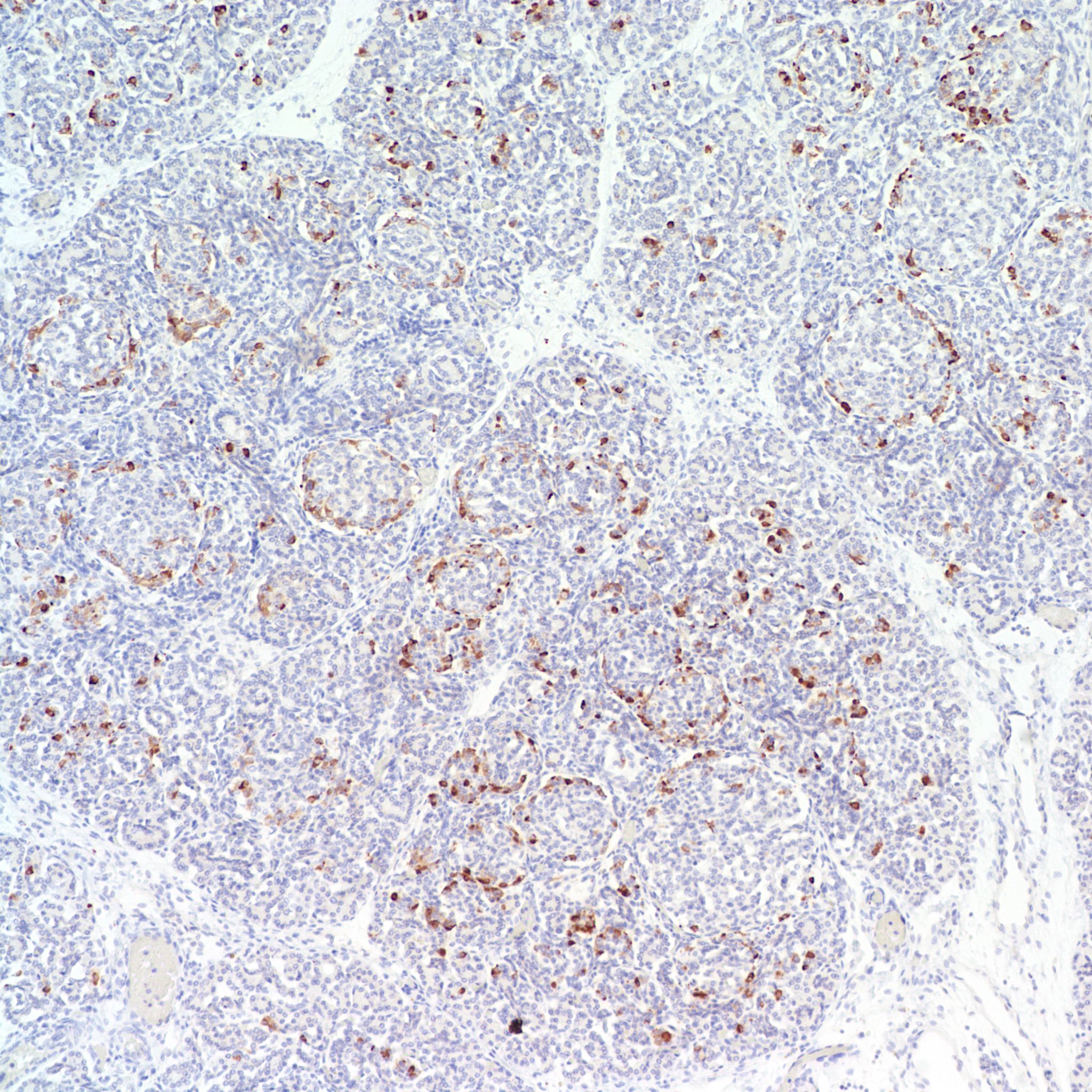
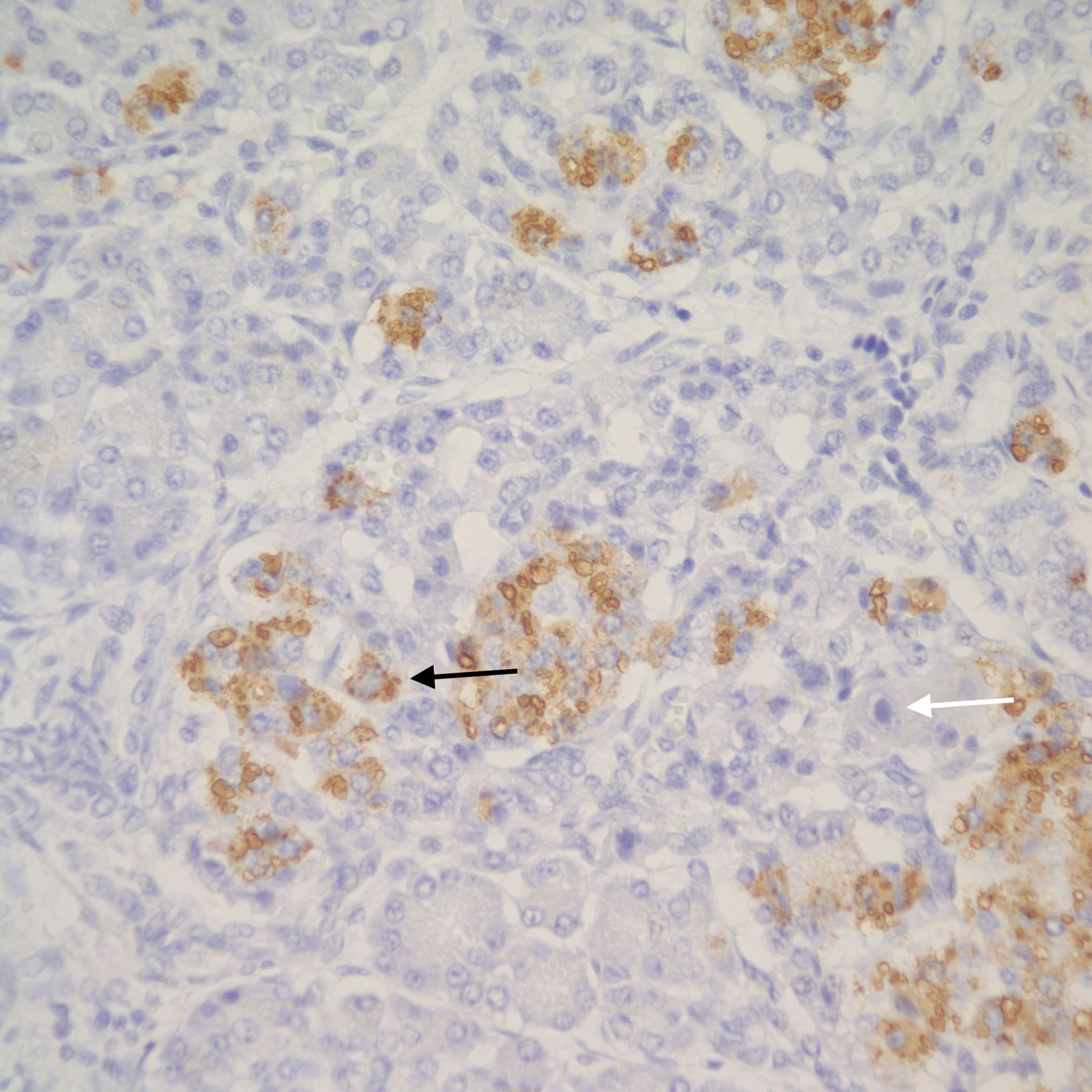
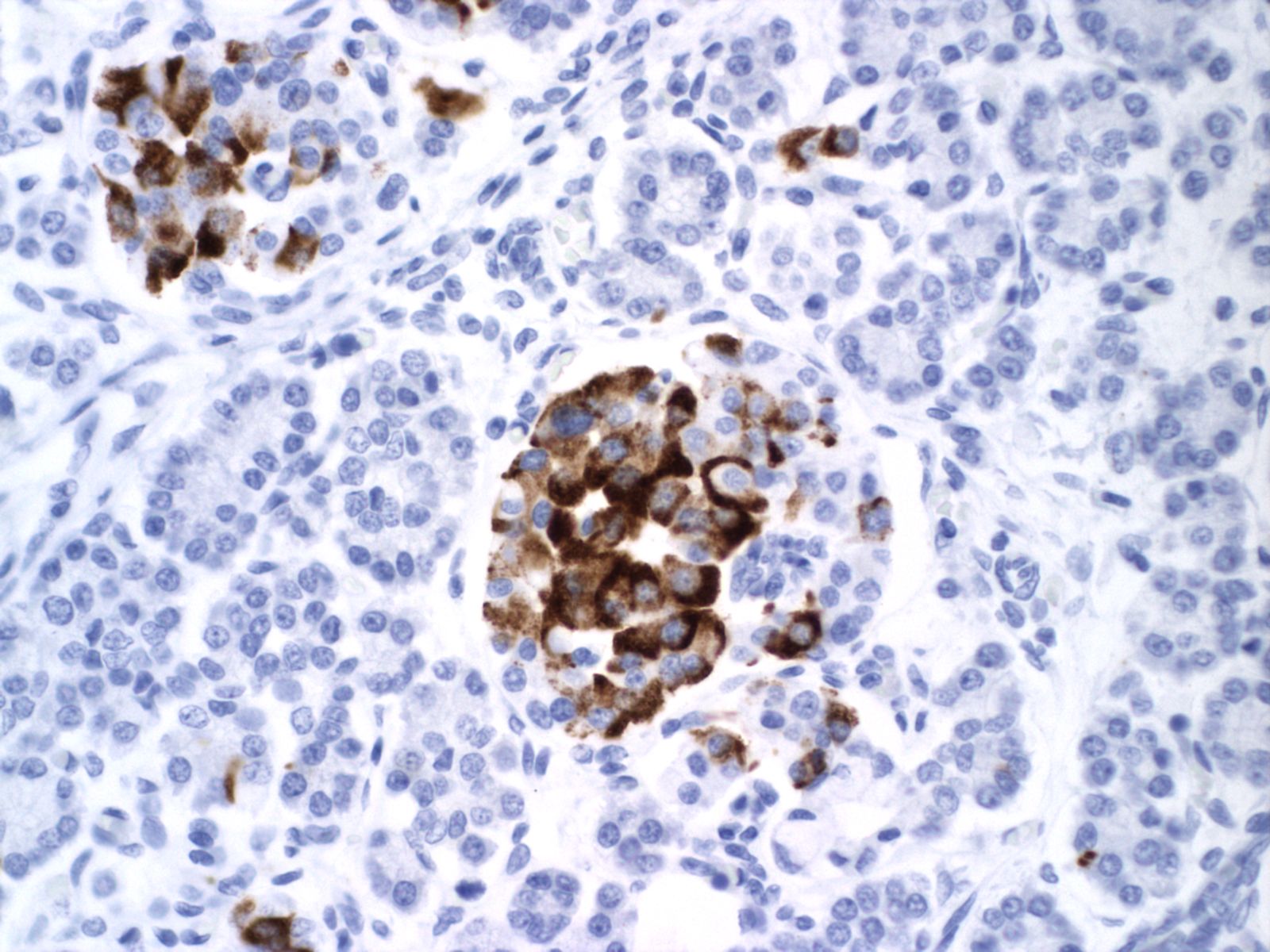
The physiological significance of the islet hyperplasia in these various diseases is unknown. Living infants of diabetic mothers often have a transient neonatal hypoglycemia presumably as a result of excess insulin secreting islet cells. In the perinatal lethal cases listed above, death precludes identifying hypoglycemia. It is also unknown what, if any, molecular mechanisms these other associations with islet hyperplasia share with the nesidioblastosis found in persistent neonatal hypoglycemia. These hypoglycemic syndromes have identifiable mutations of which at least one produces diffuse islet hyperplasia. In the Noonan-polyvalvular phenotype (Fig 15) the histology closely resembles this diffuse nesidioblastosis. A gene mutation SUR –1 mutation (Sulfonylurea receptor type 1) is also associated with hyperinsulinemia and islet cell hyperplasia[12, 13] .
Somatostatin
Small for gestational age infant’s umbilical cord blood demonstrates lower glucose and insulin levels than in appropriate for gestation infants of the same gestation[14]. In a published case of twin to twin transfusion syndrome, the smaller twin had hypertrophic islet cells that stained only for somatostatin [15]. Somatostatin in cord blood is increased in vaginally versus caesarean-delivered infants, suggesting that stress may acutely elevate the hormone [16]. Somatostatin islet cytomegaly has been reported in growth restricted fetal triploidy[17]. As with insulin, anti-somatostatin staining can identify the hormone secretion by cytomegalic cells (Fig 22).

Eosinophils
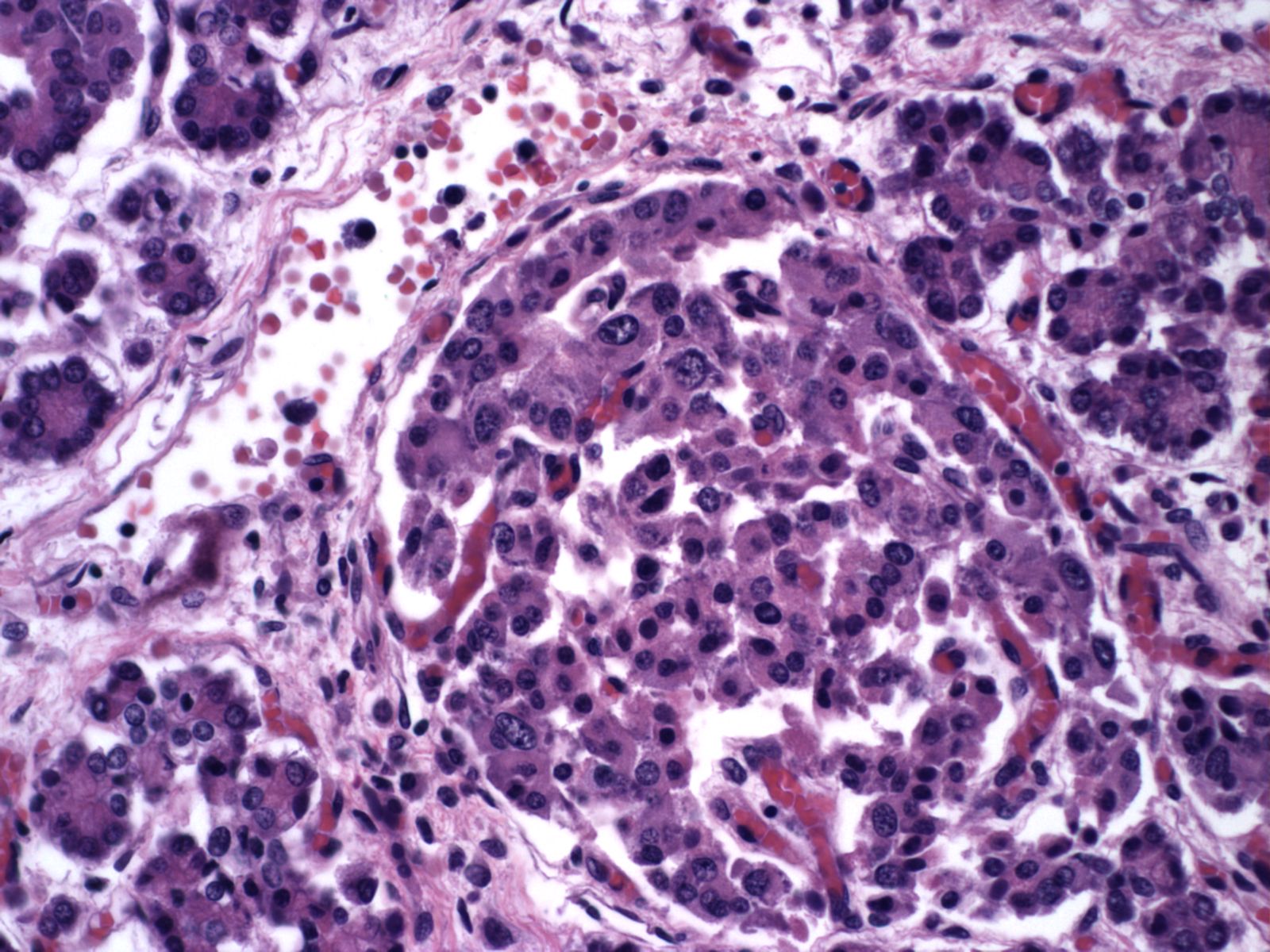
With islet hypertrophy, there is sometimes an infiltrate of predominantly eosinophils in the islet[18, 19]. Van Assche has asserted that this is evidence of type I diabetes in the mother [9]. Based on models of autoimmune diabetes, the possibility exists that the eosinophils are reducing the number of insulin cells, even as those cells must functionally increase. In a mouse model of immune diabetes, alpha islet cells (glucagon producing) were decreased in the adult mice consistent with damage to islet cells[20]. Eosinophils in the islets are also seen in pancreases with non-diabetic associations with islet hyperplasia, such as the Noonan phenotype described above (Fig 23).
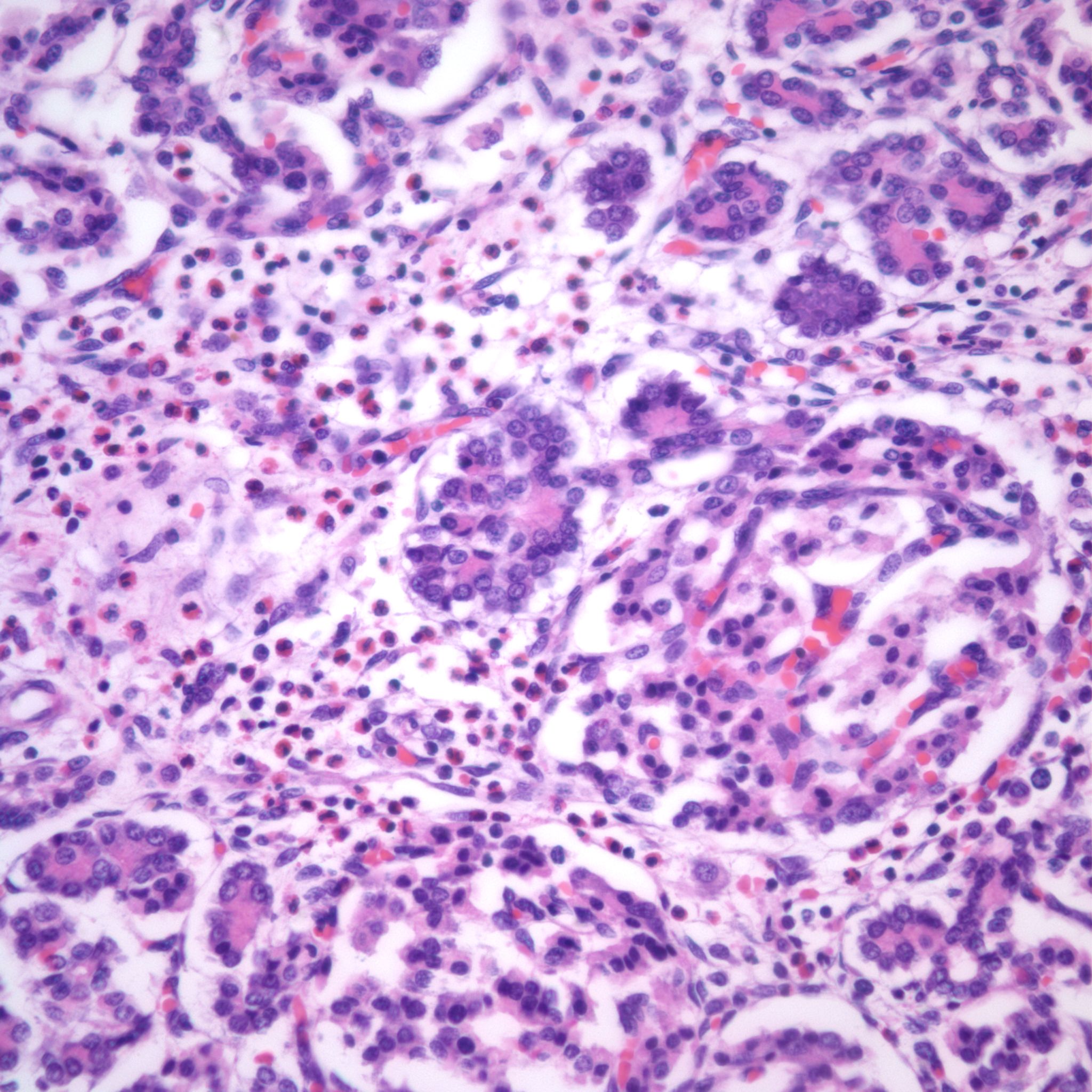
Necrobiosis of islets
The pancreas in shock shows “necrobiosis” of the islet cells, that is the nuclei become pyknotic and the cytoplasm eosinophilic (Fig 24). This lesion was identified as a lesion of cardiogenic shock in children dying of untreated hypoplastic left heart syndrome[21]. There is no report of an infant resuscitated from shock with a pancreatic polyendocrine deficiency.

Interpretation of islet findings
Directly relating the cause of death in the infant to the pancreatic findings is likely complex. Changes in the mother related to either hypoglycemia or ketoacidosis could be factors in the immediate cause of death. The relationship of the islet changes to diabetic control of the pregnancy also may not be a direct relationship. Long term changes of increased substrate such as cardiomegaly and other metabolic stresses may not be directly related to islet changes at the time of death, however, these other features may contribute to fetal death if an added stress such as acute hypoxia is superimposed. In conditions other than maternal diabetes in which increase maternal substrate is not a factor, the significance of the pancreatic findings is even less clear.
The opposite issue of low maternal substrate as a factor in many forms of growth restriction is also difficult to determine from the islet morphology. Glucagon cell mass has not yet been demonstrated to be significant. Somatostatin many be increased in some forms of stress, but the significance in terms of mortality is not clear.
An important scientific question is whether islet cell changes have any bearing on the development of insulin intolerance in later life of infants of mothers with glucose intolerance, or on the development of long-term disease in fetal growth restriction. A review article of the long term effects of fetal growth restriction reviewed epidemiologic data on birthweight and disease, and experimental models [22]. One conclusion is that elevated fetal corticosteroid may be an important factor in modifying adult susceptibility to diabetes. Experimental sheep studies suggest that many factors are involved in suppression of beta with fetal growth restriction in the sheep[23]. Somatostatin may be one of the factors, but anatomic correlates of increased secretion may be difficult to identify without measurements of somatostatin-stained cells in islets, and perhaps in the gastrointestinal tract. The interaction of stress (cortisol), glucose, insulin, and somatostatin on intrauterine islet morphology is not fully delineated. Other islet hormones have not been studied in detail in the perinatal autopsy.
As with any anatomic finding at autopsy, islet hypertrophy must be integrated with other information from the maternal history, and with other anatomic information. Prospectively planned postmortem studies using molecular methods and morphometry with comparison to experimental studies may add value above routine observations.
Infection:
The pancreas is a preferred site of involvement in some infections including congenital CMV infection (Fig 25)[24] and Coxsackie virus. Mumps is also reported to involve the pancreas.

Protocol
- Photo of posterior organ block with spleen and pancreas exposed
- Routine Histological sections
- Immunostaining for insulin, somatostatin, glucagon,
- Record relative percentages of pancreatic area and of average islet area and area of each hormone producing cell.
- Record cell hypertrophy, and hormonal staining of these cells
- Record eosinophils in and around islets.
- Record islet necrobiosis extent
- Record other lesions, such as CMV, erythroblasosis, hemosiderosis, and non-eosinophil leukocyte infiltrations
References:
1. McGeoh, J.E.M. and E.M. Darmady, Polycystic disease of kidney, liver, and pancreas. A possible pathogenesis.J Pathol, 1975. 119: p. 221-228.
2. Bernstein, J., et al., Renal-hepatic-pancreatic dysplasia: A syndrome reconsidered. Am J Med Genet, 1987. 26: p. 391-403.
3. Bendon, R.W., Ivemark’s renal-hepatic-pancreatic dysplasia: analytic approach to a perinatal autopsy. Pediatr Dev Pathol, 1999. 2(1): p. 94-100.
4. Scheele, P.M., R.W. Bendon, and M.B. Ray, Carcinoembryonic antigen, alpha 1-antitrypsin, and alpha-fetoprotein in the pancreas of patients with cystic fibrosis. Arch Pathol Lab Med, 1989. 113(10): p. 1142-6.
5. Becroft, D.M., Fetal megakaryocytic dyshemopoiesis in Down syndrome: association with hepatic and pancreatic fibrosis. Pediatr Pathol, 1993. 13(6): p. 811-20.
6. Gilson, T.P. and R.W. Bendon, Megakaryocytosis of the liver in a trisomy 21 stillbirth. Arch Pathol Lab Med, 1993. 117(7): p. 738-9.
7. Oliver, M., A. Wilson, and L. Kapila, Acute Pancreatitis and Gastric Volvulus Occurring in a Congenital Diaphragmatic Hernia. Journal of Pediatric Surgery 25: 12 (DEC 1990). 1240-1241.
8. Cardell, B., Hypertrophy and hyperplasia of the pancreatic islets in new-born infants. J Pathol Bact, 1953. 66: p. 335-346.
9. Van Assche, F.A., L. Aerts, and F.A. De Prins, The fetal endocrine pancreas. Eur J Obstet Gynecol Reprod Biol, 1984. 18(5-6): p. 267-72.
10. Pinar, H., T. Pinar, and D.B. Singer, Beta-cell hyperplasia in macrosomic infants and fetuses of nondiabetic mothers. Pediatr Dev Pathol, 2000. 3(1): p. 48-52.
11. Bendon, R. and A. Asamoah, Perinatal autopsy findings in three cases of jugular lymphatic obstruction sequence and cardiac polyvalvular dysplasia. Pediatr Dev Pathol, 2007: p. 1.
12. de Lonlay-Debeney, P., et al., Clinical features of 52 neonates with hyperinsulinism. N Engl J Med, 1999. 340(15): p. 1169-75.
13. Stanley, C.A. and L. Baker, The causes of neonatal hypoglycemia [editorial; comment]. N Engl J Med, 1999. 340(15): p. 1200-1.
14. Economides, D., A. Proudler, and K.H. Nicolaides, Plasma insulin in appropriate- and small-for-gestation-age fetuses. Am J Obstet Gynecol, 1989. 160: p. 1091-4.
15. Pietrantoni, M., et al., Mortality conference: twin-to-twin transfusion [clinical conference]. J Pediatr, 1998. 132(6): p. 1071-6.
16. Marchini, G., et al., Fetal and maternal plasma levels of gastrin, somatostatin and oxytocin after vaginal delivery and elective cesarean section. Early Human Development, 1988. 18: p. 73-79.
17. Rowlands, C.G. and W.S. Hwang, Cytomegaly of pancreatic D cells in triploidy. Pediatr Pathol Lab Med, 1998. 18(1): p. 49-55.
18. Barresi, G., C. Inferrera, and F. De Luca, Eosinophilic pancreatitis in the newborn infant of a diabetic mother.Virchows Arch A Pathol Anat Histol, 1978. 380(4): p. 341-8.
19. McKay, D., K. Benirschke, and G. Curtis, Infants of diabetic mothers histologic and histochemical observations on the pancres. Obstet Gynecol, 1953. 2: p. 133-138.
20. Pechhold, K., et al., Dynamic changes in pancreatic endocrine cell abundance, distribution, and function in antigen-induced and spontaneous autoimmune diabetes. Diabetes, 2009. 58(5): p. 1175-84.
21. Coen, R. and A.J. McAdams, Visceral manifestation of shock in congenital heart disease. Am J Dis Child, 1970. 119(5): p. 383-9.
22. Reynolds, R.M. and D.I.W. Phillips, Long-term consequences of intrauterine growth retardation. Horm Res, 1998. 49 (suppl 2): p. 28-31.
23. Kelly, A.C., et al., RNA Sequencing Exposes Adaptive and Immune Responses to Intrauterine Growth Restriction in Fetal Sheep Islets. Endocrinology, 2017. 158(4): p. 743-755.
24. Becroft, D.M.O., Prenatal cytomegalovirus infection: epidemiology, pathology, and pathogenesis. Persp Pediatr Pathol, 1981. 6: p. 203-235.
.
